| Похожие рефераты | Скачать .docx |
Курсовая работа: Органические полупроводники
Содержание
Введение
1 Полупроводниковые материалы
1.1 Общие сведения о полупроводниках
1.2 Классификация полупроводников
1.3 Собственная проводимость полупроводников
1.4 Примесная проводимость полупроводников
2 Органические полупроводники
2.1 Общая характеристика группы органических полупроводников
2.2 Характеристика отдельных групп органических полупроводников
2.3 Электропроводность органических полупроводников
2.4 Электропроводность низкомолекулярных органических полупроводников
2.5 Электрические свойства полимерных полупрводников
2.6 Механизм электропроводности
2.7 Фотопроводимость органических полупроводников
2.8 Практическое применение органических полупроводников
Экспериментальная часть
Заключение
Список литературы
Физика полупроводников , раздел физики , в котором исследуются электрические, оптические, магнитные, тепловые и другие свойства полупроводниковых материалов – широкого класса неорганических и органических веществ – и структур на их основе. Свойства полупроводников сильно зависят от внешних воздействий, а также наличия атомов примеси и собственных дефектов структуры (кристал ли чес кой решетки) . С открытия Фарадеем в 1833 г. полупроводниковых свойств у Ag2 S их отличительным признаком остается увеличение концентрации носителей заряда при нагревании, которое приводит к уменьшению электрического сопротивления материала. В отличие от металлов (проводников электричества) для полупроводников характерна чувствительность к свету (фото про води мость , люминесценция ), электрическому полю (не линей ные электрические свойства, электрический пробой ), ионизирующему излучению (радиа ци он ная физика ) и др. Полупроводники оптимально сочетают чувствительность к внешним воздействиям и возможность контролируемого формирования в них элементов с различающимися свойствами. Благодаря этому физика полупроводников служит научным фундаментом для опто- , микро- и наноэлектроники , во многом определяющих технический прогресс современного общества.
Изучение органических полупроводников вызывает сейчас наибольший интерес, так как с данными исследованиями связаны многие перспективные разработки, такие как создание OLED-дисплеев, светочувствительных материалов (например, для процессов записи информации), в микроэлектронике, для изготовления различного рода датчиков. Исследование полупроводников органических важно для понимания процессов преобразования и переноса энергии в сложных физико-химических системах и, в особенности в биологических тканях. С полупроводниками органическими, в частности с ион-радикальными солями, связана перспектива создания сверхпроводников с высокой критической температурой.
Таким образом, рассмотрение в качестве темы курсовой работы «Органические полупроводники» является актуальным.
Объектом исследования являются органические полупроводники. Предметом исследования являются конкретные свойства органических полупроводников.
Для решения поставленной цели необходимо решить следующие задачи:
- дать общую характеристику класса проводников, рассмотрев их классификацию, собственную и примесную проводимость;
- дать характеристику класса органических полупроводников, привести характеристику отдельных соединений относящихся к данному классу, рассмотреть особенности электропроводимости органических полупроводников.
- рассмотреть перспективы практического применения класса органических полупроводников, экспериментальные разработки в данной области.
С целью достижения вышеуказанных целей произвести анализ научно-методической литературы.
1.1 Общие сведения о полупроводниках
К классу полупроводников обычно относят большую группу твердых тел, удельная проводимость которых при комнатной температуре (T=300K) изменяется в очень широких пределах.
Числовое значение этой величины: (10-13 -10-1 1/Oм. см) значительно выше, чем у изоляторов: (10-26 -10-14 1/Oм. см), но намного ниже, чем у металлов: (1-102 1/Oм. см).
Если твердые тела классифицировать по механизму электропроводности, то нетрудно установить, что между полупроводниками и изоляторами не существует принципиального различия. Характерной особенностью полупроводников., отличающей их от металлов, является возрастание электропроводности s с ростом температуры, причём, как правило, в достаточно широком интервале температур возрастание происходит экспоненциально:
= 0 ехр (-EA /кТ ).(1.1.)
Здесь k — Больцмана постоянная, EA — энергия активации электронов в полупроводниках., (s0 — коэффициент пропорциональности (в действительности зависит от температуры, но медленнее, чем экспоненциальный множитель). С повышением температуры тепловое движение разрывает связи электронов, и часть их, пропорциональная exp (—EA /kT ), становится свободными носителями тока.
Связь электронов может быть разорвана не только тепловым движением, но и различными внешними воздействиями: светом, потоком быстрых частиц, сильным электрическим полем и т.д. Поэтому для полупроводников характерна высокая чувствительность электропроводности к внешним воздействиям, а также к содержанию примесей и дефектов в кристаллах, поскольку во многих случаях энергия EA для электронов, локализованных вблизи примесей или дефектов, существенно меньше, чем в идеальном кристалле данного полупроводника. Возможность в широких пределах управлять электропроводностью полупроводников изменением температуры, введением примесей и т. д. является основой их многочисленных и разнообразных применений.
Различают собственную и примесную проводимости полупроводников.
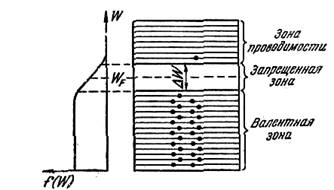
Рис.1
Важнейшее свойство полупроводников - возможность изменять свою электрическую проводимость под воздействием различных факторов: температуры, освещения, радиоактивного излучения и др.
Полупроводники представляют собой весьма многочисленный класс материалов. В него входят сотни самых разнообразных веществ – как элементов, так и химических соединений. Полупроводниковыми свойствами могут обладать как неорганические, так и органические вещества, кристаллические и аморфные, твердые и жидкие, немагнитные и магнитные. Несмотря на существенные различия в строении и химическом составе, материалы этого класса роднит одно замечательное качество- способность сильно изменять свои электрические свойства под влиянием небольших внешних энергетических воздействий. Одна из возможных схем классификации полупроводниковых материалов приведена на рис.2.
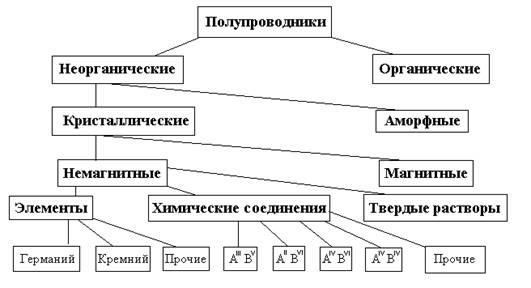
Рис. 2. Классификация полупроводниковыхматериалов по составу и свойствам.
Различие между полупроводниками и диэлектриками является скорее количественным, чем качественным. Формула (1.1) относится в равной мере и к диэлектрикам, электропроводность которых может стать заметной при высокой температуре. Точнее было бы говорить о полупроводниковом состоянии неметаллических веществ, не выделяя полупроводники в особый класс, а к истинным диэлектрикам относить лишь такие, у которых в силу больших значений EA и малых s0 электропроводность могла бы достигнуть заметных значений только при температурах, при которых они полностью испаряются.
1.2 Классификация полупроводников
Однако термин «Полупроводники» часто понимают в более узком смысле, как совокупность нескольких наиболее типичных групп веществ, полупроводниковые свойства которых четко выражены уже при комнатной температуре (300 К). Примеры таких групп:
1) Элементы IV группы периодической системы элементов Менделеева германий и кремний , которые как полупроводники пока наиболее полно изучены и широко применяются в полупроводниковой электронике . Атомы этих элементов, обладая 4 валентными электронами, образуют кристаллические решётки типа алмаза с ковалентной связью атомов, Сам алмаз также обладает свойствами полупроводника, однако величина EA для него значительно больше, чем у Ge и Si, и поэтому при Т = 300 К его собственная (не связанная с примесями или внешними воздействиями) электропроводность весьма мала.
2) Алмазоподобные полупроводники. К ним относятся соединения элементов III группы периодической системы (Al, Ga, In) с элементами V группы (Р, As, Sb), называются полупроводниками типа AIII BV (GaAs, InSb, GaP, InP и т.п.). Атомы III группы имеют 3 валентных электрона, а V группы — 5, так что среднее число валентных электронов, приходящееся на 1 атом, в этих соединениях равно 4 (как и у Ge и Si). Каждый атом образует 4 валентные связи с ближайшими соседями, в результате чего получается кристаллическая решётка, подобная решётке алмаза с той лишь разницей, что ближайшие соседи атома AIII — атомы BV а соседи атома BV — атомы AIII . За счёт частичного перераспределения электронов атомы AIII и BV в такой структуре оказываются разноимённо заряженными. Поэтому связи в кристаллах AIII BV не полностью ковалентные, а частично ионные. Однако ковалентная связь в них преобладает и определяет структуру, в результате чего эти кристаллы по многим свойствам являются ближайшими аналогами Ge и Si.
Соединения элементов II и VI групп периодической системы — AII BVI (ZnTe, ZnSe, CdTe, CdS и т.п.) также имеют в среднем 4 валентных электрона на 1 атом, но ионная связь у них более сильно выражена. У некоторых из них ковалентная связь преобладает над ионной, у других она слабее, но и те и другие обладают свойствами полупроводников, хотя и не столь ярко выраженными, как в предыдущих группах.
Представление о «средней четырёхвалентности» и «алмазоподобных» полупроводниках оказалось плодотворным для поиска новых полупроводников, например типа AII BIV C2 V (ZnSnP2 , CdGeAs2 и т.п.). Многие из алмазоподобных полупроводников образуют сплавы, которые также являются полупроводниками, например Ge — Si, GaAs — GaP и др.
3) Элементы VI и V групп и их аналоги. Элементы VI группы Te и Se как полупроводники были известны раньше, чем Ge и Si, причём Se широко использовался в выпрямителях электрического тока и фотоэлементах . Элементы V группы As, Sb и Bi — полуметаллы , по свойствам близкие к полупроводникам, а их ближайшие аналоги — соединения типа AIV и BVI (PbS, PbTe, SnTe, GeTe и т.п.), имеющие в среднем по 5 валентных электронов на атом, образуют одну из наиболее важных групп полупроводников, известную в первую очередь применением PbS, PbSe и PbTe в качестве приёмников инфракрасного излучения. Вообще среди соединений элементов VI группы (О, S, Se, Te) с элементами I—V групп очень много полупроводников. Большинство из них мало изучены. Примером более изученных и практически используемых могут служить Cu2 O (купроксные выпрямители) и Bi2 Te3 (термоэлементы).
4) Соединения элементов VI группы с переходными или редкоземельными металлами (Ti, V, Mn, Fe, Ni, Sm, Eu и т.п.). В этих полупроводниках преобладает ионная связь. Большинство из них обладает той или иной формой магнитного упорядочения (ферромагнетики или антиферромагнетики). Сочетание полупроводниковых и магнитных свойств и их взаимное влияние интересно как с теоретической точки зрения, так и для многих практических применений. Некоторые из них (V2 O3 , Fe3 O4 , NiS, EuO и др.) могут переходить из полупроводникового состояния в металлическое, причём превращение это происходит очень резко при изменении температуры.
Органические полупроводники. Многие органические соединения также обладают свойствами полупроводников. Их электропроводность, как правило, мала (s ~ 10-10 ом-1 см-1 ) и сильно возрастает под действием света. Однако некоторые органические полупроводники. (кристаллы иполимеры на основе соединений тетрацианхинодиметана TCNQ, комплексы на основе фталоцианина, перилена, виолантрена и др.) имеют при комнатной температуре s, сравнимую с проводимостью хороших неорганических полупроводников.
Для изготовления полупроводниковых приборов используют как монокристаллы, так и поликристаллические материалы. Монокристаллы представляют собой более простые системы, с более совершенным строением, чем поликристаллические материалы. Они наиболее глубоко изучены, физические явления в них лучше поддаются расчетам, и они обеспечивают большую надежность и идентичность параметров полупроводниковых приборов.
В механизме электропроводности аморфных неорганических и кристаллических органических полупроводников выявлен ряд особенностей. Интерес к органическим полупроводникам вызван тем, что в некоторых из них полупроводниковые свойства сочетаются с эластичностью, которая позволяет изготавливать рабочие элементы в виде гибких лент и волокон.
1.3 Собственная проводимость полупроводников
Собственная проводимость. Собственная проводимость возникает в результате перехода электронов с верхних уровней валентной зоны в зону проводимости. При этом в зоне проводимости появляется некоторое число носителей тока — электронов, занимающих уров ни вблизи дна зоны; одновременно в валентной зоне освобождается такое же число мест на верхних уровнях. Такие свободные от электронов места на уровнях заполненной при абсолютном нуле валентной зоны называют дырками.
Распределение электронов по уровням валентной зоны и зоны проводимости определяется функцией Ферми. Вычисления показывают, что уровень Ферми лежит точно посредине запрещенной зоны (рис.1). Следовательно, для электронов, перешедших в зону проводимости, величина W—WF мало отличается от половины ширины запрещенной зоны. Уровни зоны проводимости лежат на хвосте кривой распределения. Поэтому вероятность их заполнения электронами можно находить по формуле
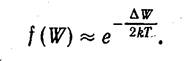 (1.2.)
(1.2.)
Количество электронов, перешедших в зону проводимости, будет пропорционально вероятности (1.2.). Эти электроны, а также образовавшиеся в таком же числе дырки, являются носителями тока.
Поскольку, проводимость пропорциональна числу носителей, она также должна быть пропорциональна выражению (1.2). Следовательно, электропроводность полупроводников быстро растет с температурой, изменяясь по закону
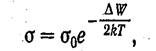 (1.3)
(1.3)
где ΔW—ширина запрещенной зоны.
Если на графике откладывать зависимость 1n σ от 1/T, то для полупроводников получается прямая линия, изображенная на рис. 2. По наклону этой прямой можно определить ширину запрещенной зоны ΔW.
Типичными полупроводниками являются элементы IV группы периодической системы Менделеева — германий и кремний. Они образуют решетку, в которой каждый атом связан ковалентными (парно-электронными) связями с четырьмя равноотстоящими от него соседними атомами. Условно такое взаимное расположение атомов можно представить в виде плоской структуры, изображенной на рис. 4. Кружки со знаком «+» обозначают положительно заряженные атомные остатки (т. е. ту часть атома, ко торая остается после удаления валентных электронов), кружки со знаком «—» — валентные электроны, двойные линии—ковалентные связи.
При достаточно высокой температуре тепловое дви жение может разорвать отдельные пары, освободив один электрон (такой случай показан на рис. 4).
Покинутое электроном место перестает быть нейтральным, в его окрестности возникает избыточный положи тельный заряд + е — образуется дырка. На это место может перескочить электрон одной из соседних пар. В результате дырка начинает также странствовать по кристаллу, как и освободившийся электрон.
Если свободный электрон встретится с дыр кой, они рекомендуют (соединяются). Это означает, что электрон нейтрализует избы точный положительный заряд, имеющийся в окрестности дырки, и теряет свободу передвижения до тех пор, пока снова не получит от кристаллической решетки энергию, достаточную для своего высвобождения. Рекомбинация приводит к одновременному исчезновению свободного электрона я дырки. На схеме уровней процессу рекомбинации соответствует переход электрона из зоны проводимости на один из свободных уровней валентной зоны.
Итак, в полупроводнике идут одновременно два процесса: рождение попарно свободных электронов и дырок и рекомбинация, приводящая к попарному исчезновению электронов и дырок. Вероятность первого процесса быстро растет с температурой. Вероятность рекомбинации пропорциональна как числу свободных электронов, так и числу дырок. Следовательно, каждой температуре соответствует определенная - равновесная концентрация электронов и дырок, величина которой изменяется с температурой по такому же закону, как и σ [см. формулу (1.2)].
В отсутствие внешнего электрического поля электроны проводимости и дырки движутся хаотически. При включении поля на хаотическое движение накладывается упорядоченное движение: электронов против поля и дырок — в направлении поля. Оба движения — и дырок, и электронов — приводят к переносу заряда вдоль кристалла. Следовательно, собственная электропроводность обусловливается как бы носителями заря да двух знаков— отрицательными электронами и положительными дырками.
Собственная проводимость наблюдается во всех без исключения полупроводниках при достаточно высокой температуре.
1.4 Примесная проводимость полупроводников
Этот вид проводимости возникает, если некоторые атомы данного полупроводника заменить в узлах кристаллической решетки атома ми, валентность которых отличается на единицу от валентности основных атомов. На условно изображена решетка германия с приме сью 5-валентных атомов фосфора. Для образования ковалентных связей с соседями атому фосфора достаточно четырех электронов. Следовательно, пятый валентный электрон оказывается как бы лишним и легко отщепляется от атома за счет энергии теплового движения, образуя странствующий свободный электрон. В отличие от рассмотренного раньше случая образование свободного электрона не сопровождается нарушением ковалентных связей, т. е. образованием дырки. Хотя в окрестности атома примеси возникает избыточный положи тельный заряд, но он связан с этим атомом и перемещаться по решетке не может. Благодаря этому заряду атом примеси может захватить приблизившийся к нему электрон, но связь захваченного электрона с атомом бу дет непрочной и легко нарушается вновь за счет тепловых колебаний решетки.
Таким образом, в полупроводнике с 5-валентной примесью имеется только один вид носителей тока — электроны. Соответственно говорят, что такой полупроводник обладает электронной проводимостью или является полупроводником n-типа (от слова negativ — отрицательный). Атомы примеси, поставляющие электроны проводимости, называются донорами.
Примеси искажают поле решетки, что приводит к возникновению на энергетической схеме так называемых локальных уровней, расположенных в запрещен ной зоне кристалла (рис. 6). Любой уровень валентной зоны или зоны проводимости может быть занят электроном, находящимся в любом месте кристалла.
Энергию, соответствующую локальному уровню, электрон может иметь, лишь находясь вблизи атома примеси, вызвавшего появление этого уровня. Следовательно, электрон, занимающий примесный уровень, локализован вблизи атома примеси.
Если донорные уровни расположены недалеко от потолка валентной зоны, они не могут существенно повлиять на электрические свойства кристалла. Иначе обстоит дело, когда расстояние таких уровней от дна зоны проводимости гораздо меньше, чем ширина запрещенной зоны, В этом случае энергия теплового движения даже при обычных температурах оказывается достаточной для того, чтобы перевести электрон с донорного уровня в зону проводимости. На этому процессу соответствует отщепление пятого валентного электрона от атома примеси. Захвату свободного электрона атомом примеси соответствует на рис. 6 переход электрона из зоны проводимости на один из донорных уровней.
Уровень Ферми в полупроводнике n-типа лежит между донорными уровнями и дном зоны проводи мости, при невысоких температурах — приблизительно посредине между ними
На условно изображена решетка кремния с примесью 3-валентных атомов бора. Трех валентных электронов атома бора недостаточно для образования связей со всеми четырьмя соседями. Поэтому одна из связей окажется неукомплектованной и будет представлять собой место, способное захватить электрон. При переходе на это место электрона одной из соседних пар возникнет дырка, которая будет кочевать по кристаллу. Вблизи атома примеси возникнет избыточный отрицательный заряд, но он будет связан с данным атомом и не может стать носителем тока. Таким образом, в полупроводнике с 3-валентной примесью возникают носители тока только одного вида — дырки. Проводимость в этом случае называется дырочной, а о полупроводнике говорят, что он принадлежит к p-типу (от слова positiv — положительный). Примеси, вызывающие возникновение дырок, называются акцепторными.
На схеме уровней акцептору соответствует расположенный в запретной зоне недалеко от ее дна локальный уровень. Образованию дырки отвечает переход электрона из валентной зоны на акцепторный уровень. Обратный переход соответствует разрыву одной из четырех ковалентных связей атома примеси с его соседями и рекомбинации образовавшегося при этом электрона и дырки.
Уровень Ферми в полупроводнике р-типа лежит между потолком валентной зоны и акцепторными уровнями, при невысоких температурах — приблизительно посреди не между ними.
С повышением температуры концентрация примесных носителей тока быстро достигает насыщения. Это означает, что практически освобождаются все донорные или заполняются электронами все акцепторные уровни. Вместе с тем по мере роста температуры все в большей степени начинает сказываться собственная проводимость полупроводника, обусловленная переходом электронов непосредственно из валентной зоны в зону проводимости. Таким образом, при высоких температурах проводимость полупроводника будет складываться из примесной и собственной проводимости. При низких температурах преобладает примесная, а при высоких — собственная проводимость.
2.1 Общая характеристика группы органических полупроводников
Полупроводники органические , твёрдые органические вещества, которые имеют (или приобретают под влиянием внешних воздействий) электронную или дырочную проводимости. Полупроводники органические характеризуются наличием в молекулах системы сопряжения. Носители тока в полупроводниках органических образуются в результате возбуждения p-электронов, делокализованных по системе сопряжённых связей. Энергия активации, необходимая для образования носителей тока в полупроводниках органических, снижается по мере увеличения числа сопряжений в молекуле и в полимерах может быть порядка тепловой энергии.
К полупроводникам органическим относятся органические красители (например, метиленовый голубой, фталоцианины), ароматические соединения (нафталин, антрацен, виолантрен и др.), полимеры с сопряжёнными связями, некоторые природные пигменты (хлорофилл, b-каротин и др.), молекулярные комплексы с переносом заряда, а также ион-радикальные соли. Полупроводники органические существуют в виде монокристаллов, поликристаллических или аморфных порошков и плёнок. Величины удельного сопротивления r при комнатной температуре у полупроводников органических лежат в диапазоне от 1018 ом ×см (нафталин, антрацен) до 10-2 ом ×см . Наиболее проводящими полупроводниками органическими являются ион-радикальные соли, на основе анион-радикала тетрацианхинодиметана. Они обнаруживают электропроводность металлического характера. У полупроводников органических с низкой электропроводностью наблюдается явление фотопроводимости.
Полупроводники органические обладают особенностями, которые определяются молекулярным характером их структуры и слабым межмолекулярным взаимодействием:
1) поглощение света вызывает возбуждение молекул, которое может мигрировать по кристаллу в виде экситонов;
2) образование носителей тока под действием света связано с распадом экситонов на поверхности кристалла, дефектах его структуры, примесях, при взаимодействии экситонов друг с другом, а также с автоионизацией высоковозбуждённых молекул;
3) зоны проводимости узки (~0,1 эв ), подвижность носителей тока, как правило, мала (~1 см2 /в ×сек );
4) наряду с зонным механизмом электропроводности осуществляется прыжковый механизм.
В кристаллах ион-радикальных солей межмолекулярное взаимодействие сильно анизотропно, что приводит к высокой анизотропии оптических и электрических свойств и позволяет рассматривать этот класс полупроводники органические как квазиодномерные системы.
Полупроводники органические находят применение в качестве светочувствительных материалов (например, для процессов записи информации), в микроэлектронике, для изготовления различного рода датчиков. Исследование полупроводников органических важно для понимания процессов преобразования и переноса энергии в сложных физико-химических системах и в особенности в биологических тканях. С полупроводниками органическими, в частности с ион-радикальными солями, связана перспектива создания сверхпроводников с высокой критической температурой.
2.2 Характеристика отдельных групп органических полупроводников
1. Красители, цветные органические соединения, применяемые для окраски текстильных материалов, кожи, мехов, бумаги, пластмасс, резин, древесины и др. К ним относятся также бесцветные соединения, из которых окрашенные вещества образуются после нанесения на материал, например красители для холодного крашения, а также отбеливатели оптические. Природные красители — ализарин, индиго и др. — добывались с глубокой древности из растений, реже из животных организмов. Первые синтетические красители получены в 1856 независимо польским химиком Я. Натансоном (фуксин) и английским химиком У. Г. Перкином (мовеин), а в 1857 начато промышленное производство мовеина. В 1869 синтезирован ализарин (немецкие химиками К. Гребе и К. Т. Либерманом) и вскоре большое число др. синтетических красителей, превосходящих по качеству природные. К началу 20 в. синтетические красители почти полностью вытеснили натуральные. Синтез красителей стал возможным после открытия Н. Н. Зининым общего метода получения ароматических аминов. К началу 70-х гг. число красителей, выпускавшихся промышленностью всего мира, превышало 9000 и ежегодно растет.
По химическому строению красители разделяют на следующие группы: нитрокрасители, нитрозокрасители, азокрасители, арилметановые, хинониминовые красители, сернистые красители, индигоидные красители, антрахиноновые красители, полициклические красители, фталоцианиновые красители, полиметиновые красители, азометиновые красители. По областям и методам применения красители делят на кислотные, прямые, кубовые, сернистые, протравные, основные, катионные, активные (реактивные), окислительные, дисперсные, пигменты и лаки, жиро-, спирто- и ацетонорастворимые, красители для холодного (ледяного) крашения, для кожи, алюминия, меха, дерева и др.
Цветность красителей, т. е. способность избирательно поглощать видимые лучи света, связана с их химическим строением: наличием достаточно протяжённой системы сопряжённых двойных связей, часто включающей гетероатомы. На окрашиваемом материале (субстрате) красители удерживаются благодаря образованию химических связей с субстратом: ковалентных (в случае активных красителей) или ионных (для кислотных красителей), а также силами адсорбции и водородной связи (прямые красители); многие красители образуют нерастворимые в воде частицы (кубовые, сернистые красители, красители для холодного крашения), которые «застревают» в порах субстрата; для удержания красители на окрашиваемом материале применяют также связующие или плёнки полимера. В процессе эксплуатации материала его окраска не должна существенно изменяться под действием света, слабых кислот и щелочей, при стирке, трении, глаженье и т. п. Устойчивость окраски зависит от многих факторов, в том числе от химического строения красителя, характера связи красителя с субстратом и природы последнего. Так, например, основные красители нестойки на шерсти, но достаточно прочны на полиакрилонитрильном волокне. Устойчивость окрасок к различным воздействиям измеряется по пятибалльной системе, кроме светопрочности, которая оценивается по восьмибалльной шкале.
Помимо окраски различных материалов, красители применяют в цветной и черно-белой кинематографии и фотографии, аналитической химии, в медицине в качестве средств диагностики, при биохимических исследованиях, в жидкостных лазерах, в различных физических приборах в качестве элементов, обладающих фотопроводимостью и некоторыми др. свойствами.
Сырьём для производства красителей служат бензол, нафталин, антрацен, пирен и др. ароматические и гетероциклические соединения, а также различные кислоты, щёлочи, соли, спирты и др. вещества. Сначала получают так называемые промежуточные продукты, которые далее превращают в красители посредством реакций конденсации, диазотирования, азосочетания, окисления и др. производство многих красителей отличается сложностью, процесс иногда состоит из 10 и более стадий.
2. Хлорофилл (от греч. chlorós — зелёный и phýllon — лист), зелёный пигмент растений, с помощью которого они улавливают энергию солнечного света и осуществляют фотосинтез. Локализован в особых клеточных структурах — хлоропластах или хроматофорах и связан с белками и липидами мембран. Основу структуры молекулы Х, составляет магниевый комплекс порфиринового цикла; в IV пиррольном кольце к остатку пропионовой кислоты присоединён высокомолекулярный спирт фитол, который придаёт Х. способность встраиваться в липидный слой мембран хлоропластов.
Высшие растения и зелёные водоросли содержат Х. а и в , бурые и диатомовые водоросли — а и с , красные водоросли — Х. а и d. В фотосинтезирующих бактериях присутствуют близкие аналоги Х. — бактериохлорофиллы. По своему строению Х. близок к др. природным комплексам порфиринов (с железом) — дыхательным пигментам — цитохромам, красящему веществу крови — гему, а также простетическим группам некоторых ферментов — пероксидазы, каталазы.
Название «Х.» было дано французскими химиками П. Пельтье и Ж. Каванту зелёному спиртовому раствору смеси растительных пигментов в 1817. Впервые Х. а и в разделил в начале 20 в. русским учёный М. С. Цвет с помощью разработанного им хроматографического метода. Химическую структуру Х. выяснили немецкие учёные Р. Вильштеттер,А. Штоль (1913), Х. Фишер (1930—40). Полный синтез Х. осуществил американский химик Р. Вудворд. Роль Х. в фотосинтезе доказана классическими работами К.А. Тимирязева. Пути биосинтеза Х. выяснены в трудах американских учёных Д. Шемина, С. Граника и др.; большой вклад в изучение Х. внесли советские учёные Т.Н. Годнев и А.А. Шлык.
Основной путь биосинтеза Х. определяется конденсацией двух молекул d-аминолевулиновой кислоты с образованием порфобилиногена — производного пиррола, который в результате ряда ферментативных превращений даёт соединение, содержащее порфириновое ядро — протопорфирин IX. Из протопорфирина образуется непосредственный предшественник Х. — протохлорофиллид, уже содержащий атом магния. Путём последующих реакций восстановления и присоединения фитола из этого предшественника образуется Х. Стадия восстановления протохлорофиллида осуществляется у высших растений на свету, у низших растений — в темноте.
В хлоропластах и хроматофорах большая часть Х. (содержание его обычно составляет 0,5—1,5% на сухую массу) находится в виде светособирающей «антенны» и меньшая часть — в реакционных центрах, непосредственно участвующих в работе цепи фотосинтетического переноса электрона. Поглощая квант света, молекула Х. переходит в возбуждённое состояние (длительность жизни синглетного возбуждённого состояния около 10-9 сек ), которое может переходить в долгоживущее триплетное возбуждённое состояние с длительностью жизни до 10-3 сек. Возбуждённые светом молекулы Х. способны переносить электрон от молекулы-донора к молекуле-акцептору. Механизм этих реакций в модельных системах выяснен в работах советских учёных А.А. Красновского, В.Б. Евстигнеева и др. Способность возбуждённого Х. к переносу электрона обеспечивает функционирование реакционных центров фотосистем цепи фотосинтетического переноса электрона. Применение спектральной техники и низких температур показало, что в первичном фотоакте бактериохлорофилл, а возможно, и Х. активного центра отдают свой электрон молекуле-акцептору (убихинон, ферредоксин). Этот первичный фотопроцесс сопряжён с цепью энзиматических реакций, ведущих к образованию восстановленных пиридиннуклеотидов и аденозинтрифосфата, обеспечивающих работу углеродного цикла. Т. о., свет, поглощённый Х., преобразуется в потенциальную химическую энергию органических продуктов фотосинтеза и молекулярного кислорода. Свет, поглощаемый Х., вызывает в клетках также др. фотобиологические явления: индуцирует генерацию электрического потенциала на мембранах хлоропластов, влияет на движение одноклеточных организмов (фототаксис) и т.д.
Исследованию свойств Х. на разных уровнях молекулярной организации уделяется большое внимание, т.к. эти свойства тесно связаны с фундаментальным явлением преобразования энергии света в химическую энергию при фотосинтезе.
2.3 Электропроводность органических полупроводников
Изучение неорганических и органических полупроводников показало, что вних возникают следующие виды зарядоносителей:
а) атомы, которые, потеряв свой электрон с внешней оболочки,становятся положительно заряженными частицами и участвуют в переносеположительных зарядов;
б) освобожденные при этом изменении электроны, которые становятся
носителями зарядов;
в) ионизированные атомы-акцепторы, т.е. атомы, захватившие усоседнего атома электрон; они тоже являются отрицательно заряженными
частицами и участвуют в переносе отрицательных частиц;
г) дырки, образовавшиеся при захвате у атома валентных электронов;они начинают притягивать электроны от соседнего атома и становятсясвоеобразными носителями положительного электричества.
Значительно больше видов движения зарядоносителей у органических полупроводников. Здесь их перемещение представляет собой совмещение сложных
явлений, одно из которых обусловлено «блуждающими» по молекуле электронами.Так как молекулы различны, то и связи их с электроном различны.
Электрическое поле, в которое помещен полупроводник, вызывает направленное движение носителей (дрейф), обусловливающее протекание тока в полупроводнике.
Основным для круга вопросов, связанных с прохождением электрического тока в полупроводнике, является понятие подвижности носителей m, определяемое, как отношение средней скорости направленного их движения (скорости дрейфа), вызванного электрическим полем uд , к напряжённости Е этого поля:
m = uд /Е (2.1)
Подвижности разных типов носителей в одном и том же полупроводнике различны, а в анизотропных полупроводниках различны и подвижности каждого типа носителей для разных направлений поля. Дрейфовая скорость, возникающая в электрическом поле, добавляется к скорости теплового хаотического движения, не дающего вклада в ток. Тот факт, что при заданном поле носитель имеет постоянную дрейфовую скорость uд , а не ускоряется неограниченно, связан с наличием процессов торможения — рассеяния. В идеальном кристалле даже в отсутствие поля каждый носитель имел бы определённую и неизменную как по величине, так и по направлению скорость uд . Однако реальный кристалл содержит примеси и различные дефекты структуры, сталкиваясь с которыми носитель каждый раз меняет направление скорости — рассеивается, так что движение его становится хаотическим. Под действием поля носитель эффективно ускоряется только до момента очередного столкновения, а затем, рассеиваясь, теряет направленность своего движения и энергию, после чего ускорение в направлении поля Е начинается заново до следующего столкновения. Т. о., средняя скорость его направленного движения набирается только за интервал времени Dt между 2 последовательными столкновениями (время свободного пробега) и равна:
uд = eE Dt/m,
откуда:
m = -е Dt/т. (2.2.)
Процессы рассеяния носителей тока разнообразны. Наиболее общим для всех веществ является рассеяние на колебаниях кристаллической решётки (фононах), которые вызывают смещения атомов кристалла из их положений равновесия в решётке и тем самым также нарушают её упорядоченность. Испуская или поглощая фононы, носитель изменяет свой квазиимпульс, а, следовательно, и скорость, т. е. рассеивается. Средняя частота столкновений 1/ Dt зависит как от природы кристалла, интенсивности и характера его колебаний и содержания в нём примесей и дефектов, так и от энергии носителей. Поэтому m зависит от температуры. При температурах T ~ 300 К определяющим, как правило, является рассеяние на фононах. Однако с понижением температуры вероятность этого процесса падает, т.к. уменьшается интенсивность тепловых колебаний решётки, а, кроме того, малая тепловая энергия самих носителей позволяет им испускать не любые возможные в данном кристалле фононы, а лишь небольшую часть из них, имеющих достаточно малые энергии (частоты). В таких условиях для не очень чистых кристаллов преобладающим становится рассеяние на заряженных примесях или дефектах, вероятность которого, наоборот, растет с понижением энергии носителей.
Представления о свободном движении носителей, лишь изредка прерываемом актами рассеяния, применимы, однако, лишь к полупроводникам с не слишком малым m (m ³ 1 см2 /сек ). Для меньшей подвижности l становится меньше размеров элементарной ячейки кристалла (~10-8 см ) и теряет смысл, т.к. само понятие «свободного» движения носителей в кристалле связано с переходом их из одной ячейки в другую (внутри каждой ячейки электрон движется, как в атоме или молекуле). Столь малые значения m характерны для многих химических соединений переходных и редкоземельных металлов с элементами VI группы периодической системы элементов и для большинства полупроводников органических. Причиной является, по-видимому, сильное взаимодействие носителей с локальными деформациями кристаллической решётки, проявляющееся в том, что носитель, локализованный в какой-либо элементарной ячейке, сильно взаимодействуя с образующими её и соседние ячейки атомами, смещает их из тех положений, которые они занимают, когда носителя нет. Энергия носителя в такой деформированной ячейке (поляроне) оказывается ниже, чем в соседних недеформированных, и переход его в соседнюю ячейку требует затраты энергии, которую он может получить от какой-либо тепловой флуктуации. После перехода покинутая носителем ячейка возвращается в недеформированное состояние, а деформируется та, в которую он перешёл. Поэтому следующий его переход в 3-ю ячейку снова потребует энергии активации и т. д. Такой механизм движения называется прыжковым, в отличие от рассмотренного выше зонного, связанного со свободным движением носителей в разрешенных зонах и не требующего затраты энергии на переход из ячейки в ячейку. При прыжковом механизме не имеют смысла такие представления зонной теории твёрдого тела, как квазиимпульс, эффективная масса, время и длина свободного пробега, но понятия средней скорости дрейфа под действием поля и подвижности остаются в силе.
Прыжковый механизм электропроводности характерен для многих аморфных и жидких полупроводников. Носители с энергиями в области псевдозапрещённой зоны переходят от состояния локализованного вблизи одной флуктуации к другой путём таких активированных перескоков (т. к. энергии состояний вблизи разных флуктуаций различны, поскольку сами флуктуации случайны и по расположению и по величине). В полупроводниках с высокой подвижностью иногда при низких температурах также наблюдается прыжковая проводимость (если подавляющее большинство носителей локализовано на примесях, они могут перескакивать с примеси на примесь). Явления переноса в полупроводниках с малой подвижностью пока поняты в меньшей мере, чем для полупроводников с зонным механизмом проводимости.
2.4 Электропроводимость низкомолекулярных органических полпроводников
Важнейшие сведения о процессах, лежащих в основе электропроводности вещества, дают измерения ее температурной зависимости. Такие измерения, проведенные различными авторами на большом числе объектов, показали, что для чистых органических веществ температурная зависимость электропроводности в охваченных интервалах температур может быть описана соотношением (1.1):
Так, при перескоковом механизме энергия активации будет определяться из выражения с экспонентой ![]() , при зонном механизме проводимости энергия активации входит в экспоненту
, при зонном механизме проводимости энергия активации входит в экспоненту ![]() . В большинстве работ по низкомолекулярным органическим полупроводникам принято определять термическую энергию активации из последнего выражения.
. В большинстве работ по низкомолекулярным органическим полупроводникам принято определять термическую энергию активации из последнего выражения.
Такая зависимость установлена у большого числа органических соединений, принадлежащих самым различным классам. Были исследованы антрацен, нафталин, полициклические ароматические соединения, азо-ароматичеекие соединения, органические пигменты фталоцианины, гематин, гематопорфирин, хлорофилл, органические красители трифенилметанового, цианинового, ксантенового, акридинового и других классов, радикал α, α-дифенилпикрилгидразил, каротин, бензофенон, некоторые кислоты, фталимиды.
Низкомолекулярные органические соединения являются, как правило, весьма высокоомными проводниками, и измерения электропроводности их могут быть проведены лишь при достаточно высоких температурах. В таких случаях величина проводимости при комнатной температуре определяется экстраполяцией прямых:
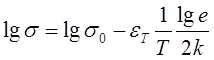 (2.3)
(2.3)
Для большинства соединений удельная проводимость при комнатной температуре составляет 10-10 -10-20 ом-1 ·см-1 .
В то время как сам экспоненциальный характер зависимости проводимости от обратной температуры, по-видимому, не вызывает сомнений, значения энергии активации ![]() ,определяемые для одних и тех же веществ разными авторами, часто оказываются резко различными. Как показывает сопоставление таких данных, величина
,определяемые для одних и тех же веществ разными авторами, часто оказываются резко различными. Как показывает сопоставление таких данных, величина ![]() может зависеть от условий приготовления исследуемого образца, от его структуры (аморфный слой, полукристаллический порошок или монокристалл), от условий предварительной его обработки, от того, исследовалась ли электропроводность в вакууме, в атмосфере газа или на воздухе.
может зависеть от условий приготовления исследуемого образца, от его структуры (аморфный слой, полукристаллический порошок или монокристалл), от условий предварительной его обработки, от того, исследовалась ли электропроводность в вакууме, в атмосфере газа или на воздухе.
Такой разброс значений ![]() , безусловно, связан с тем, что некоторые методы приготовления образцов и подготовки их к измерениям не обеспечивают должной их чистоты (наличие примесей, адсорбированных газов или влаги). Поэтому в целом ряде случаев удавалось наблюдать лишь примесную проводимость, несмотря на то, что для приготовления образца использовалось вещество, казалось бы, достаточно высокой степени чистоты, и условия опыта были также достаточно совершенны (например, глубокая эвакуация, термическая обработка образца в вакууме и т. п.). Так, для натертого на кварц слоя фталоцианина меди Вартанян получил при измерениях в вакууме значение
, безусловно, связан с тем, что некоторые методы приготовления образцов и подготовки их к измерениям не обеспечивают должной их чистоты (наличие примесей, адсорбированных газов или влаги). Поэтому в целом ряде случаев удавалось наблюдать лишь примесную проводимость, несмотря на то, что для приготовления образца использовалось вещество, казалось бы, достаточно высокой степени чистоты, и условия опыта были также достаточно совершенны (например, глубокая эвакуация, термическая обработка образца в вакууме и т. п.). Так, для натертого на кварц слоя фталоцианина меди Вартанян получил при измерениях в вакууме значение ![]() =0,9 эв. Многочасовая тренировка слоя в вакууме при температуре 100° С сопровождалась падением электропроводности слоя и ростом энергии активации до значения 1,2 эв. Было установлено, что эти изменения обусловливались постепенной десорбцией кислорода, поскольку напускание кислорода на оттренированный слой вновь приводило к уменьшению энергии активации. То, что даже наивысшая энергия активации, полученная в этих опытах, оставалась много меньше энергии оптического перехода, соответствующей положению участка длинноволнового спада полосы поглощения (1,6—1,8 эв), привело Вартаняна к выводу, что наблюдаемая в этих условиях проводимость была примесной. Зависимость величины проводимости и энергии активации от степени обезгаженности слоев указывала на то, что примесью служил адсорбированный на красителе кислород, который при откачке и примененной термообработке полностью не удалялся.
=0,9 эв. Многочасовая тренировка слоя в вакууме при температуре 100° С сопровождалась падением электропроводности слоя и ростом энергии активации до значения 1,2 эв. Было установлено, что эти изменения обусловливались постепенной десорбцией кислорода, поскольку напускание кислорода на оттренированный слой вновь приводило к уменьшению энергии активации. То, что даже наивысшая энергия активации, полученная в этих опытах, оставалась много меньше энергии оптического перехода, соответствующей положению участка длинноволнового спада полосы поглощения (1,6—1,8 эв), привело Вартаняна к выводу, что наблюдаемая в этих условиях проводимость была примесной. Зависимость величины проводимости и энергии активации от степени обезгаженности слоев указывала на то, что примесью служил адсорбированный на красителе кислород, который при откачке и примененной термообработке полностью не удалялся.
Собственную проводимость фталоцианина меди с энергией активации 1,7 эв Вартанян и Карпович наблюдали на слоях, полученных возгонкой в условиях высокого вакуума. Этот метод не является единственным, который для фталоцианинов обеспечивает высокую чистоту образца. В работе Филдинага и Гутмена исследовались монокристаллы фталоцианина меди и также была установлена собственная проводимость (![]() =1,64 эв). Собственную проводимость с энергией 1,79 эв удалось найти и для поликристаллических порошков фталоцианина меди, Освобождение вещества от адсорбированного кислорода достигалось в этом случае путем нагревания до 340° С. Измерения проводились в потоке азота.
=1,64 эв). Собственную проводимость с энергией 1,79 эв удалось найти и для поликристаллических порошков фталоцианина меди, Освобождение вещества от адсорбированного кислорода достигалось в этом случае путем нагревания до 340° С. Измерения проводились в потоке азота.
Все это свидетельствует о том, что если образцы органического полупроводника получаются таким методом или подвергаются такой обработке, которые обеспечивают наибольшую их чистоту, то независимо от метода получения образцов и от характера их структуры наблюдается собственная проводимость с характерной для данного вещества энергией активации.
При перемещении носителей тока в процессе проводимости путь их складывается из участков движения внутри молекул и участков межмолекулярного переноса. Очевидно, что для аморфных, поликристаллических образцов и монокристаллов условия движения носителей между молекулами будут различны. Поэтому близость энергий активации для них указывает на определяющую для электропроводности роль внутримолекулярных процессов. В то же время было показано, что могут наблюдаться различия в величине проводимости у соединений, для которых π-электронные системы тождественны, но условия межмолекулярного переноса электронов различны. Фалмайер и Вольф сопоставили электропроводность обычного и хлорированного фталоцианинов меди. В последнем 15 из 16 атомов водорода замещены атомами хлора. Было найдено, что энергии активации для этих соединений весьма близки (1,79 и 1,86 эв соответственно), но проводимость у хлорированного фталоцианина на порядок выше. Это свидетельствует о том, что наличие тяжелых концевых атомов (в данном случае атомов хлора) обеспечивает лучшие условия для перехода носителей между молекулами, и это приводит к росту подвижности носителей. При неоднократных нагреваниях хлорированного соединения до температур 400—500°С, с которых велось измерение температурной зависимости проводимости, молекулы его постепенно теряли хлор, и прямые  сохраняя наклон, приближались к прямой для исходного фталоцианина меди.
сохраняя наклон, приближались к прямой для исходного фталоцианина меди.
2.5 Электрические свойства полимерных полупрводников
Электрические характеристики полимеров с сопряженными связями имеют широкий диапазон значений: от диэлектрических до полу металлических.
Удельная электропроводность колеблется от 10-19 до единиц Ом-1 ·см-1 ; энергия активации проводимости — от 2 до 0,01 эв. Обратимая температурная зависимость электропроводности носит типичный полупроводниковый характер: α экспоненциально растет с температурой по закону Аррениуса.
Отсутствие поляризационных эффектов при длительном пропускании тока свидетельствует о том, что проводимость имеет, как правило, электронную, а не ионную природу.
Исследования полупроводниковых полимеров показывают, что закон Ома для многих образцов соблюдается до напряженности поля 103 —104 в/см, хотя иногда отмечаются отклонения уже в весьма малых полях— порядка 10—100 в/см. Знак носителей тока, определяемый по термо-эде. может быть как положительным (дырки), так и отрицательным (электроны). Однако кислород всегда изменяет термо-эде в положительную сторону, и поэтому возможно, что в противоположность первоначальным представлениям об органических полупроводниках носителями тока в являются преимущественно электроны (как это следует из большинства измерений к вакууме), а дырочная проводимость— результат адсорбции кислорода. У ряда полимерных проводников отмечен внутренний фотоэффект.
У некоторых полимерных полупроводников отмечена способность выпрямлять электрический ток. т. с. неомическое поведение у контактов, что позволило осуществить n-р-переход. Интересно, однако, что из некоторых полимеров удалось получить материалы с истинным р-n-переходом, в частности из радиационно и термически обработанного полиэтилена, который спрессовывали со слоями йодированного полимера, имеющего иной тип проводимости. Коэффициент выпрямления такого р-n-контакта доходил до 25.
Электропроводность полимерных полупроводников с повышением температуры увеличивается примерно па 1—3% на 1 °С. электропроводность неорганических полупроводников на 3—6%. В противоположность электропроводности металлов, которая уменьшается с повышением температуры примерно на 0,3% на 1 °С.
Однако значение температурного коэффициента электропроводности полимеров-полупроводников в большей степени зависит от температурной области, в которой проводятся определения. При очень низких температурах температурный коэффициент можег достигать 20—40% на 1 °С.
Полимерные полупроводники, как и промышленные, обнаруживают термо-э.д.с. в пределах 3 300 мв /град и более.
Подвижность носителей тока в полимерных полупроводниках очень низкая 0,005—0,04 см2 /(в·сек) и не может быть измерена с помощью эффекта Холла. Только в пирополимерах отмечается подвижность носителей тока, достигающая 2—100 см2 /(в·сек). У неорганических полупроводников подвижность носителей тока составляет 200— 400 см2 /(в·сек) и более, хотя у неорганических окислов, например у NiO. подвижность постелей тока мала. По-видимому, высокой подвижности носителей тока можно достичь главным образом на монокристаллических образцах, у которых нет диэлектрических прослоек.
2.6 Механизм электропроводности
Объяснение процессов переноса тока в органических полупроводниках, и особенно в полимерах, пожалуй, наиболее серьезная проблема, возникающая при изучении этих интересных веществ. Здесь следует различать два вопроса: во-первых, как зарождаются носители тока и, во-вторых, каким образом они перемещаются в объеме твердого тела. Механизм возникновения носителей в сопряженных системах не вызывает особых трудностей для понимания. Зарождение носителей тока должно достаточно легко происходить на участках полимера с высокой степенью сопряжения, поскольку с ростом числа сопряженных связей снижается внутримолекулярный барьер для переброса электрона на свободные уровни молекулы и. следовательно, ослабевает возбуждение электрона при переходе в квазисвободное проводящее состояние внутри молекулы. Действительно, опытами по измерению проводимости на переменном токе показано, что энергия зарождения носителей внутри области сопряжения близка к нулю и уж во всяком случае значительно меньше энергии межмолекулярных переходов.
Гораздо сложнее вопрос о механизме перемещения носителей заряда между сопряженными молекулами или областями сопряжения, поскольку именно этой стадией лимитируется суммарный процесс электропроводности во всем объеме полимера. В любом предлагаемом механизме должен учитываться ряд установленных сейчас особенностей, которые необычны не только для органических веществ, но и для большинства хорошо изученных неорганических полупроводников. Рассмотрим важнейшие из таких особенностей.
Электронная неоднородность . Измерения разными методами (исследования на переменном токе; изучение шумов тока, влияния адсорбции иода и воздуха и др.) позволили прийти к выводу о микрогетерогенности структуры полупроводниковых полимеров, т.е. о том, что все полимеры состоят из отдельных хорошо проводящих областей (по всем вероятности, сильно сопряженных или конденсированных участков), разделенных плохо проводящими диэлектрическими участками, очевидно с неупорядоченной структурой полимерных молекул.
Характер обратимой температурной зависимости термо-эдс. В большинстве случаев коэффициент термо-эдс α или почти не зависит от температуры, или растет при нагревании. Это означает, что температурная зависимость α так же как температурная зависимость σ определяется изменением подвижности носителей при их постоянной концентрации, так как при таком механизме теория предсказывает слабый (логарифмический) рост α с увеличением температуры:
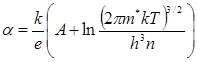 ,(2.4)
,(2.4)
где А — константа, зависящая от механизма рассеяния электронов; ![]() и n — эффективная масса и концентрация носителей тока;
и n — эффективная масса и концентрация носителей тока; ![]() и h - постоянные Больцмана и Планка; Т-абсолютная температура. Такие проводники называют вырожденными.
и h - постоянные Больцмана и Планка; Т-абсолютная температура. Такие проводники называют вырожденными.
В случае, когда изменение значений α и σ в зависимости от температуры обусловлено главным образом экспоненциальным ростом концентрации носителей (невырожденные проводники), α будет согласно теории падать с ростом температуры пропорционально величине 1/T:
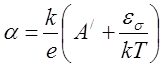 .(2.5)
.(2.5)
Для некоторых полимеров обнаружено снижение α с температурой, однако и в этих случаях обычно имеет место снижение недостаточно сильное для того, чтобы его можно было отнести за счет определяющего влияния концентрации носителей.
Очень малая подвижность носителей заряда. Попытки измерить подвижность носителей в полимерах с помощью эффекта Холла оказывались, как правило, неудачными. Это означает, что подвижность носителей не превышает 0.005—0.01 см /в·сек. Лишь в нескольких случаях такие измерения дали положительные результаты: например, для одного из полиацепхинонных радикалов было получено очень малое значение подвижности: u = 0.04 см2 /в·сек, а для комплекса поливинилкарбазола с иодом u = 0,4 см2 /в·сек. В полифталоцианине меди обнаружилась неожиданно высокая подвижность u = 2.5 10 см2/в·сек., однако воспроизвести измерение эффекта Холла не удалось, а подвижность, определенная по влиянию адсорбции кислорода на электропроводность, оказалась гораздо меньшей порядка 102 см2 /в·сек. Столь малым значениям подвижности соответствует концентрация носителей тока в полимерах не более 1012 -1017 в 1 см3 .
Только тогда, когда, по-видимому, устраняется влияние диэлектрических прослоек (например, в результате термообработки полимера), возможно обнаружить более высокую подвижность: от единиц до 100 см2 /в·сек.
При изучении полимерных полупроводников возникает ряд вопросов: какова природа электропроводности, чем объясняется низкая подвижность носителей тока, природа фотопроводимости и др.
Современное представление электропроводности неорганических полупроводников базируется на зонной теории.
Дли полимерных полупроводников обсуждаются перескоковый, туннельный и зонный механизмы проводимости.
Зонный механизм предполагает межмолекулярные электронные взаимодействия, приводящие к возникновению общей для всего объема полимера зоны проводимости, в которой концентрация носителей зависит от энергии вырывания электрона из сопряженной системы с переходом его в квазисвободное проводящее состояние (концентрация носителей тока зависит от ширины запрещенной зоны) и которая увеличивается с ростом температуры.
С учетом всех указанных особенностей есть несколько вариантов объяснения механизма переноса тока в органических полупроводниках. Прежде всего пробуют использовать зонный механизм, детально разработанный для неорганических полупроводников. В применении к рассматриваемым сопряженным системам зонный механизм дает следующую картину. Взаимодействия между электронами ведут к возникновению общей для всего вещества зоны проводимости, в которой концентрация носителей экспоненциально растет с ростом температуры. При этом низкая подвижность объясняется узостью зон проводимости. Однако зонные представления вряд ли могут служить основой для построения общего механизма электропроводности в полупроводниковых полимерах, поскольку такой схеме в большинстве случаев противоречат положительный температурный ход α и слишком низкая подвижность, при которой понятие «зона» теряет смысл. В целом зонный механизм неприменим. В частности, зонному механизму противоречит температурный ход термо-Э.Д.С.
Согласно другому механизму, называемому туннельным, электропроводность определяется вероятностью межмолекулярного туннельного перехода электронов, иными словами, частотой квантовомеханического без активационного «просачивания» сквозь межмолекулярный барьер. Эффективность туннелирования пропорциональна концентрации электронов на возбужденных уровнях, которая в свою очередь экспоненциально растет с увеличением температуры. В некоторых случаях туннельный механизм подтверждается совпадением экспериментальных данных с расчетными, полученными с учетом формы барьеров. Однако и этот механизм, по-видимому не является главным в случае полимерных полупроводников, потому что согласно теоретическим расчетам он "маловероятен" для веществ с низкой подвижностью носителей.
Туннельный механизм предполагает, что истинная энергия активации проводимости определяется энергией перевода электрона на возбужденный уровень, поэтому для полимеров с системой сопряженных связей энергия активации проводимости должна быть мала и с ростом степени сопряжения в макромолекуле стремиться к нулю. Эффективность туннельных переходов пропорциональна концентрации электронов на возбужденных уровнях, которая растет с температурой. Измерения электропроводности на переменном токе говорят в пользу весьма малых энергий активации зарождения носителей внутри области сопряжения (~0,1 эв).
Перескоковый механизм электропроводности предполагает возможность протекания тока посредством активационных перескоков носителей тока из одной области хорошей проводимости полимера (по-видимому, области полисопряжения) в другую с преодолением энергетических барьеров, создаваемых плохо проводящими (диэлектрическими) барьерами (неупорядочной или несопряженной структурой). Повышение температуры не изменяет числа эффективных носителей тока, возникающих в полисопряженных областях (областях «коллективного» взаимодействия я-электронов), а увеличивает вероятность перескоков, т. е. их число (подвижность носителей). Считают, что перескоковый механизм наиболее вероятен в веществах с малой подвижностью носителей тока [0,005— 0,01 см2 /(в·сек)], где эффект Холла не измеряется, хотя общая концентрация носителей может быть порядка 10 —10|8 см3 .
Пожалуй, наиболее удовлетворительное объяснение особенностей проводимости в органических полупроводниках в настоящее время можно дать с помощью перескокового механизма, согласно которому ток возникает благодаря активационным перескокам носителей из одной полисопряженной области в другую над диэлектрическими барьерами, создаваемыми неупорядоченной (несопряженной) структурой. Зарождение и перемещение носителей внутри полисопряженной области почти не требуют энергии активации. Рост температуры не изменяет концентрации носителей, а экспоненциально увеличивает вероятность перескоков, т.е. подвижность. Как показывает расчет, в системах с таким перемещением носителей наблюдать эффект Холла очень трудно. Теоретические работы последних лет указывают на большую вероятность перескокового механизма по сравнению с другими схемами для систем с низкой подвижностью носителей. В связи с этим интересно подчеркнуть, что измерениями на переменном токе удалось экспериментально подтвердить весьма малые значения энергии активации зарождения носителей внутри области сопряжения (около 0,1 эв), в то время как измерения на постоянном токе показали, что процесс электропроводности лимитируется стадией со значительной энергией активации (порядка 1,0 эв), т. е. очевидно, надбарьерными перескоками между участками сопряжения.
Таким образом, для большинства типичных полимерных полупроводников наиболее правдоподобным следует, по-видимому, признать перескоковый механизм, хотя высказано также мнение, что электрическое поведение полупроводниковых полимеров определяется наложением двух активационных процессов — изменение концентрации носителей тока и изменение их подвижности; подобный механизм предлагался ранее для низкомолекулярных органических полупроводников. В ряде случаев может оказаться пригодной и обычная зонная схема — например, когда подвижность носителей тока больше 1 см2 /в·сек. или когда α снижается с увеличением температуры.
Нужно, однако, иметь в виду, что в некоторых группах полимеров электропроводность осуществляется заведомо по иным схемам. Это имеет место, в частности, когда подвижность превышает 10 см2 /в·сек (в высокотемпературных образцах, в композициях с высоким содержанием металла или при образовании сильных КПЗ с полимерами), а также, когда обнаруживается резкое падение α с увеличением температуры. По всей вероятности, в этих случаях электропроводность имеет квазиметаллическую природу из-за сильных электронных взаимодействий в полимере. При этом концентрация носителей может возрасти до 1019 в 1 см3 .
2.7 Фотопроводимость органических полупроводников
Другая важная особенность органических полупроводников, отличающая их от большинства органических веществ,— это фотопроводимость, т.е. возрастание электропроводности при освещении объекта, фототок i растет с увеличением интенсивности L, освещения по закону:
![]() ,(2.6)
,(2.6)
где n может иметь значение от 1 (линейная люкс-амперная характеристика) до 0,5. В полях с напряжением менее нескольких киловольт на 1 см фототок i изменяется по закону Ома. Носителями фототока, как и в случае темновой проводимости (т. е. проводимости в отсутствие освещения), могут быть и электроны и дырки. Однако при проведении опыта в вакууме, тщательной очистке или уплотнении структуры слоя вещества знак носителей может изменяться с положительного (дырки) на отрицательный (электроны). Это означает, что дырочный тип проводимости часто обусловлен присутствием адсорбированного кислорода — сильного акцептора электронов, изменяющего соотношение электронов и дырок в пользу последних. В большинстве изученных веществ дырки, по-видимому, подвижнее электронов. Как правило, знак носителей фототока совпадает со знаком темновых носителей. Органические полупроводники обнаруживают фотоэлектрическую чувствительность в широком диапазоне частот спектра. Изучение спектральной зависимости фотопроводимости показало, что в очень тонких слоях или в монокристалле максимальные значения фототока совпадают с максимумами в спектрах поглощения. Однако для обычных поликристаллических объектов в области максимума поглощения наблюдается минимум фототока. Это объясняют тем, что интенсивное образование носителей фототока, происходящее под действием поглощаемого света в объеме вещества, может не вызвать возрастания фототока из-за еще более интенсивной рекомбинации (исчезновения) этих постелей, протекающей при освещении на поверхности.
Температурная зависимость фотопроводимости, так же как темповой, соответствует экспоненциальному закону:
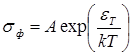 .(2.7)
.(2.7)
Однако величина термической энергии активации фотопроводимости ![]() составляет всего несколько десятых электрон-вольта и имеет тенденцию к росту по мере увеличения размеров молекулы.
составляет всего несколько десятых электрон-вольта и имеет тенденцию к росту по мере увеличения размеров молекулы.
Другое объяснение малых значений термической энергии активации фотопроводимости заключается в том, что ![]() определяется той небольшой дополнительной энергией, которая необходима для переброса носителей в зону проводимости с уровней ловушек, расположенных в запрещенной зоне вблизи дна зоны проводимости. А заброс носителей на эти уровни происходит в результате освещения вещества.
определяется той небольшой дополнительной энергией, которая необходима для переброса носителей в зону проводимости с уровней ловушек, расположенных в запрещенной зоне вблизи дна зоны проводимости. А заброс носителей на эти уровни происходит в результате освещения вещества.
Оптическую энергию активации фотопроводимости (равную половине ![]() минимальной энергии световых квантов, необходимой для возникновения фототока) определяют, например, по красной (длинноволновой) границе фоточувствительности, т.е. по той минимальной частоте световых волн, при которой появляется фототок. Как правило, получаемые этим методом данные для многих веществ близки к значениям, вычисляемым из термической энергии активации темновой проводимости
минимальной энергии световых квантов, необходимой для возникновения фототока) определяют, например, по красной (длинноволновой) границе фоточувствительности, т.е. по той минимальной частоте световых волн, при которой появляется фототок. Как правило, получаемые этим методом данные для многих веществ близки к значениям, вычисляемым из термической энергии активации темновой проводимости ![]() , и к энергии оптического возбуждения, определяемой по длинноволновой границе оптического поглощения:
, и к энергии оптического возбуждения, определяемой по длинноволновой границе оптического поглощения:
![]()
![]() (2.8)
(2.8)
Следовательно, темновая электропроводность и фотопроводимость имеют, по-видимому, общую природу, и поэтому выводы о связи электрических свойств со строением молекулы и структурой вещества и о механизмах проводимости следует относить ко всему электронному поведению этих веществ.
Помимо фотопроводимости многие низкомолекулярные органические полупроводники обнаруживают и другое фотоэлектрическое свойство фотовольтаическую активность, или фото-эдс. Она проявляется либо форме вентильной фото-эде — возникновение потенциала при освещении контакта исследуемого вещества с металлом или с другим веществом, либо в форме диффузионной фото-эдс, возникающей за счет различия в подвижности электронов и дырок, которые образуются при импульсном освещении вещества перпендикулярно обкладкам — электродам. Область спектральной чувствительности фото-эдс обычно совпадает с областью поглощения в оптических спектрах.
Многие полимеры с сопряженными связями обнаруживают фотоэлектрический эффект— рост проводимости при освещении. По своему характеру наблюдаемые при этом закономерности в общем аналогичны тем явлениям, которые хорошо изучены для соединений других классов органических полупроводников. Для большинства полимеров выявлена электронная природа фотопроводимости. Наряду с этим обнаружены некоторые аномалии, например тушение фотопроводимости кислородом в дырочных фотопроводниках и поляризационные явления. К сожалению, значительные фотоэффекты наблюдаются лишь у немногих полимеров, содержащих в цепи сопряжения тройные связи.
Большой интерес вызывает обнаруженное для некоторых полимеров явление фотосенсибилизации, т.е. усиление фотоэффектов (Фото-эдс и фотопроводимости) при введении в полимер красителей. Это явление аналогично хорошо известному фотосенсибилизирующему действию красителей на неорганические полупроводники.
Другие характерные особенности электрического поведения полупроводниковых полимеров рассмотрены в связи с механизмами проводимости. Здесь же следует указать еще на так называемый компенсационный эффект, установленный для ряда полимеров, низкомолекулярных сопряженных систем, комплексов с переносом заряда и даже диэлектриков. Заключается он в том, что между параметрами уравнения Аррениуса для электропроводности существует определенная зависимость, благодаря которой в какой-то группе веществ значения электропроводности могут мало отличаться друг от друга, так как различия в энергии активации εТ будут компенсироваться соответствующей разницей в величине предэкспоненциального множителя А согласно линейному соотношению
Такая зависимость присуща, только плохим полупроводникам и не должна соблюдаться в низкоомных полимерах.
Изучение фотоэлектрических процессов в полимерах наряду с изучением темповой электропроводности дает дополнительную информацию об электронных явлениях в таких системах, гак как световой луч позволяет зондировать определенные свойства электронов.
Многие исследования покачали, что спектры фотопроводимости и спектры фото-эдс обычно имеют большое сходство со спектрами поглощения. Например, на толстых слоях наблюдается уменьшение фотоэлектрической чувствительности в максимуме поглощения: для появления фотопроводимости необходимо наличие развитой поверхности. Носители заряда, по-видимому, образуются на поверхности фоточувствительного материала, куда диффундирует экситон.
Фотопроводимость у фоточувствительных образцов изменяется экспоненциально с ростом температуры:
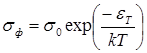 ,(2.8)
,(2.8)
где ![]() фотопроводимость;
фотопроводимость; ![]() — константа для данного образца;
— константа для данного образца; ![]() — термическая энергия активации проводимости (обычно 0,1—0,3 эв).
— термическая энергия активации проводимости (обычно 0,1—0,3 эв).
Знак световых носителей тока у большинства органических полупроводников дырочный. Некоторые адсорбированные пары и газы существенно изменяют фотоэлектрическую чувствительность органических полупроводников.
Зависимость фототока от освещенности выражается формулой:
![]() ,(2.9)
,(2.9)
где n— коэффициент 9.5—1,0; L —освещенность.
Полиацетилениды меди имеют темповое сопротивление 109 —1010 ) ом·см, которое при освещении монохроматическим светом уменьшается до 10— 108 ом·см с нарастанием фототока до максимального значения за 10—15 сек.
Сухой кислород обратимо уменьшает фотопроводимость на два-три порядка и фото-эдс, в 3 — 5 раза. Аналогично действуют gары воды.
Механизм фотоэффекта сводится к возбуждению макромолекулы поглощенным фоnоном. Возбужденное состояние мигрирует до встречи со структурным или химическим дефектом, на котором происходит образование свободной пары носителей фототока. Электрон задерживается положительно заряженными центрами, а свободные дырки мигрируют по системе макромолекул.
Существенным фактором, определяющим фотоэлектрическую чувствительность вещества, являются потенциальные барьеры на границе между макромолекулами. В связи с ним возрастает роль случайных связей, обусловливающих сшивание макромолекул.
Па полиииах и полиацетиленидах обнаружено явление спектральной сенсибилизации фотоэффекта красителями, такими, как метиленовый голубой, хлорофилл, фталоцианины, родамин и др. Подобно красителям ведет себя иод, следы которого могут увеличивать фотопроводимость.
Полученные результаты показали, что при сенсибилизации фотоэффекта органических полупроводников наблюдаются закономерности, свойственные неорганическим полупроводникам.
Высокая фотоэлектрическая чувствительность полиинов и полиацетиленидов позволила создать на их основе электрофотографические слои, чувствительность которых сравнима с чувствительностью слоев из неорганических полупроводников.
2.8 Практическое применение органических полупроводников
Рассмотрим один из аспектов применения органических полупроводников, а именно, технологию OLED (Organic Light Emitting Diode), уже в названии которой содержатся два кардинальных отличия от LCD технологии — “органический” и “светоизлучающий”.
С начала 1960-х г. микроэлектроника основывается исключительно на неорганических материалах: кремнии, германии, арсенидегаллия, металлических проводниках из алюминия или меди, различных диэлектриках типа того же диоксида кремния.
Тем не менее, все это время не прекращалась исследовательская работа по органическим материалам — полимерам и олигомерам, а также гибридным органическим — неорганическим соединениям по всему спектру параметров: проводимость, полупроводниковые качества, светоизлучение. Не говоря уже о том что органика обладает рядом интересных качеств вроде более мягких требований к температуре окружающей среды, зачастую выдающейся гибкостью и т. д., что открывает перед производителями электронных устройств ряд совершенно новых применений.
В последние годы органические материалы используются даже в производстве центральных процессоров: проводящие органические соединения применяются в упаковке процессоров, для Intel — начиная еще с OLGA (Organic Land Grid Array), и в литографии — в качестве фоторезистивных материалов.
Однако с ростом проблем, встающих сегодня перед традиционной неорганической микроэлектроникой, вероятность того, что производители начнут обращать больше внимания на органику, становится все выше и выше. Пионером в их исследовании стал Eastman Kodak — ученые Чин Тэнг и Стив Ван Слайк еще в 1987 году издали статью “Organic electroluminescent diodes”, описывающую новый класс тонкопленочных устройств на базе органических материалов, обладающих электролюминесцентными качествами, заметно превосходящими все, что было создано в этой области ранее.
Впервые предложенная Kodak схема с двумя слоями органики между электродами вместо одного и сегодня остается основным вариантом, используемым для создания OLED устройств. Все закрыто стеклом, покрытым со стороны OLED тончайшим слоем оксидa олова индия (indium tin oxid), выступающим в роли анода. Непосредственно к нему прилегает первый органический слой, порядка 750 ангстрем (75 нм) ароматического диамина, выступающего в роли полупроводника “р типа”, следом идет основной, светоизлучающий слой из пленки, состоящей из соединения, принадлежащего к классу fluorescent metal chelate комплексов. Например, hydroxyquinoline aluminium. В роли полупроводника n типа может выступать много органических соединений с труднопроизносимыми на основе все тех же ароматических углеводов. И, наконец, последним слоем является катод, состоящий из смеси магния с серебром с атомным соотношением 10:1. Эта система имеет толщину менее 500 нм, вместе с задней подсветкой, каковой она, помимо всего прочего, сама и является. При прохождении тока напряжением от 2, 5 В базовый слой начинает излучать фотоны, чей поток становится все более интенсивным по мере увеличения силы тока, усиливаясь практически линейно и позволяя при напряжении менее 10 В получить яркость более 1 000 Кд на квадратный метр, что минимум в два раза превышает соответствующий показатель LCD экранов (максимум же — свыше 100 000 Кд на квадратный метр).
Пик интенсивности спектра приходится на 550 нм длину волны, что соответствует зеленому цвету. Естественно, кроме явных плюсов были и минусы. Тут и долговечность, точнее, ее отсутствие — в первоначальных опытах светимость при постоянном напряжении падала вдвое уже после 100 часов непрерывной работы, и проблемы с отдельными участками спектра — в частности, с голубым.
Тем не менее прорыв был очевиден, учитывая, что до этого для получения более менее нормальной светимости требовалось напряжение порядка 100 В. К решению оставшихся проблем присоединилось множество фирм (на сегодняшний день OLED занимаются порядка восьмидесяти компаний и университетов), и большинство из них в той или иной мере уже можно считать решенными. Новые OLED материалы представляют куда более сложные комбинации веществ, чем это было на заре их истории. Новые химические формулы базовых слоев, отдельные обогащающие добавки, отвечающие каждая за свою часть спектра — красную, синюю, зеленую. Основные усилия разработчиков направлены в настоящий момент на улучшение характеристик органических полупроводников. Успехи более чем впечатляют: хотя в синем спектре последние перспективные OLED материалы и остаются наименее долговечными, тем не менее, даже в условиях синей светимости их срок жизни достигает 10 тысяч часов. Красный и зеленый цвета дают до 40 тысяч, универсальный белый — 20 тысяч часов. Уже прилично, учитывая, что для тех же цифровых камер, к примеру, среднее время жизни экрана считается нормальным от 1 000 часов. К тому же в коммерческих продуктах речь, очевидно, будет идти о классической схеме, используемой в LCD, когда экран состоит из сплошных белых OLED излучателей с цветными фильтрами, отвечающими за придание цвета конкретным пикселям.
Ко всему прочему новые основные материалы значительно повышают и физические параметры OLED. В частности, повышая верхнюю планку диапазона рабочих температур более чем до 100 градусов по Цельсию, с прицелом на использование в автомобильной электронике и подобных устройствах.
Как в традиционных CRT экранах, OLED экран представляет собой матрицу, состоящую из комбинаций ячеек трех основных цветов — красного, синего, зеленого.
В зависимости от того, какой цвет от него требуется, регулируется уровень напряжения на каждой из ячеек матрицы, в результате чего смешением трех получившихся оттенков и достигается требуемый результат. В своем развитии OLED экраны начали с пассивных матриц, которые прекрасно подходят, на пример, для экранов автомагнитол или дешевых сотовых телефонов. Такая матрица представляет собой простейший двухмерный массив пикселей в виде пересекающихся строк и колонок. Каждое пересечение является OLED диодом. Что бы подсветить его, управляющие сигналы подаются на соответствующие строку и колонку. Чем больше подано напряжение, тем ярче светимость пикселя. Напряжение требуется достаточно высокое, вдобавок подобная схема не позволяет создавать эффективные экраны, состоящие более чем из миллиона пикселей. Когда у первых ноутбуков курсор мыши, двигающийся по экрану, оставлял за собой длинный, угасающий след — вот это и есть пример пассивной матрицы.
Весьма схожи между собой у LCD и OLED принципы работы активной матрицы. Все тот же двухмерный массив из пересекающихся колонок и линий, но на сей раз каждое из их пересечений представляет собой не только светоизлучающий элемент, жидкокристаллическую ячейку или OLED диод, но и управляющий им транзистор. Управляющий сигнал посылается уже на него, он запоминает, какой уровень светимости от ячейки требуется, и, пока не будет дана другая команда, будет исправно поддерживать этот уровень тока. И напряжение в этом случае требуется куда ниже, и ячейка куда быстрее реагирует на изменение ситуации.
Транзисторы здесь требуются не совсем обычные — они должны лечь еще одним ровным тонким слоем на предыдущие слои. Исходя из этой задачи появился новый класс устройств — тонко пленочные транзисторы — TFT. Делались они из сугубо неорганических материалов, а именно — из того же привычного кремния. Немного другого, разумеется — гидрогенизированного аморфного кремния, за счет своей физической структуры более медленного, чем привычный нам по чипам монокристаллический кремний.
Рассмотрим более подробно свойства, лежащие в основе функционирования OLED дисплеев.
Основой для создания органических материалов особой группы – так называемых проводящих электролюминесцентных полимеров служат высокомолекулярные соединения с чередующимися двойными связями в молекулах. В чистом виде они не являются проводниками заряда, поскольку электроны в них локализованы вследствие участия в образовании сильных химических связей. Для освобождения электронов применяются различные примеси, после добавления, которых и появляется возможность перемещения зарядов (электронов и дырок) вдоль молекулярной цепи.
Таким образом, в основе подобной технологии лежат свойства так называемых сопряженных полимеров. В их молекулах атомы углерода образуют между собой двойные или тройные связи. Каждый атом выбирает партнера-фаворита, чтобы отдавать ему два электрона вместо обычного одного. "Лишний" электрон делится еще с одним соседом-атомом. В результате перекрытия p-орбиталей появляются "свободные" электроны и, как следствие, становится возможным протекание электрического тока вдоль молекулярных цепей. Возникают энергетические зоны валентности и проводимости, разделенные запретной зоной. Так полимеры приобретают свойства полупроводников. Эти материалы обладают теми же свойствами, что и неорганические полупроводники, то есть, способны образовывать p-n–переход и, что особенно важно, при определенных условиях излучать свет. Это позволило создать комбинированные по принципу действия устройства – излучающие диоды.
В исследованиях OLED выделилось два основных направления, одно из которых заложили ученые из Eastman-Kodak. Опубликовав еще в 1987 г. статью Organic Electroluminiscent Diodes, они описали новый класс тонкопленочных устройств на базе органических материалов с электролюминесцентными свойствами, заметно превосходящими все, что было создано в этой области ранее. Предложенная Kodak схема с двумя слоями органики между электродами вместо одного и сегодня остается одним из основных вариантов, используемых для создания OLED-устройств. При этом технологический процесс использует циклы вакуумного испарения (осаждения). В феврале 1999 г. корпорации Sanyo Electric и Eastman-Kodak образовали альянс для разработки и продвижения на рынке OLED-дисплеев. Уже через несколько месяцев они смогли показать работающий прототип полноцветного активно-матричного дисплея.
Другое направление – Polymer LED (PLED) – было заложено в 1989 г., когда профессор Ричард Френд (Richard Friend) вместе с группой химиков научной лаборатории Кембриджского университета открыл светоизлучающие полимеры LEP (Light Emitting Polymer). Вскоре выяснилось, что открытые вещества обладают рядом свойств, которые позволяют разработать на их основе семейство дисплеев нового поколения. Для изучения LEP и создания новых дисплеев была образована компания CDT. Вскоре она нашла инвесторов, и началась разработка первого дисплея, сделанного на основе LEP- или PLED-технологии.
Специалистам из CDT удалось решить ряд проблем, применив, например, специальные методики по производству упорядоченных полимеров, а также использовав новые материалы. Чтобы добиться излучения света, был спроектирован аналог неорганического диода. Он состоял из двух слоев – полифениленвинилена (polyphenylene-vinylene, PPV) и циано-PPV (CN-PPV), размещенных между полупрозрачным электродом (окислы индия и олова), который наносили на подложку стекла, с одной стороны, и металлическим контактом – с другой. Эти материалы – PPV и циано-PPV – являются не только полупроводниками, но и, кроме того, еще и самоизолирующими полимерами. Как показали ученые, CN-PPV хорошо подходит для транспортировки электронов благодаря более низкому положению дна зоны проводимости. Электрические характеристики материалов подобраны так, чтобы электроны из CN-PPV и дырки из PPV собирались вдоль границы контакта слоев, где и происходит их рекомбинация с генерацией фотонов.
На сегодняшний день OLED/PLED-технологиями занимаются несколько десятков компаний и университетов. Новые материалы представляют собой куда более сложные комбинации веществ, чем было возможно на заре этих технологий: новые химические формулы базовых слоев, отдельные обогащающие добавки, отвечающие каждая за свою часть спектра – красную, синюю и зеленую. Ведь как и в традиционных ЭЛТ-дисплеях, OLED-экран представляет собой матрицу, состоящую из комбинаций ячеек трех основных цветов – красного, синего, зеленого. В зависимости от того, какой цвет требуется получить, регулируется уровень напряжения на каждой ячейке матрицы, в результате чего смешением трех образовавшихся оттенков и получается искомый результат.
Итак, структура OLED-ячейки многослойна. Сверху OLED-панели располагается металлический катод, снизу – прозрачный анод. Между ними находится несколько органических слоев, собственно и составляющих светодиод. Один слой является источником дырок, второй – полупроводниковым каналом, третий слой транспортирует электроны и, наконец, в четвертом слое происходит замещение дырок электронами, которое в светоизлучающих полимерах сопровождается световым излучением.
Как и ЖК, OLED-дисплеи бывают активными и пассивными. Последний тип дисплея представляет собой простейший двухмерный массив пикселов в виде пересекающихся строк и колонок. Каждое такое пересечение является OLED-диодом. Чтобы заставить его излучать свет, управляющие сигналы подаются на соответствующую строку и колонку. Чем большее подано напряжение, тем ярче будет светимость пиксела. Напряжение требуется довольно высокое, вдобавок подобная схема, как правило, не позволяет создавать большие экраны, состоящие более чем из миллиона пикселов.
Что касается активной матрицы, то это все тот же двухмерный массив из пересекающихся колонок и линий, но на сей раз каждое из их пересечений представляет собой не только светоизлучающий элемент (или OLED-диод), но и управляющий им тонкопленочный транзистор. Управляющий сигнал посылается уже на него, а он, в свою очередь, "запоминает", какой уровень светимости от ячейки требуется, и пока не получит другую команду, будет исправно поддерживать этот уровень тока. И напряжение в таком случае требуется куда ниже, и ячейка куда быстрее реагирует на изменение ситуации. Обычно здесь используются тонкопленочные полевые транзисторы – TFT (Thin Film Transistor) на базе поликристального кремния.
Благодаря партнерству CDT с корпорацией Seiko-Epson произошло, пожалуй, важнейшее событие в истории развития пластиковых дисплеев. Японцы предложили с помощью модифицированной струйной технологии "печатать" пикселы экрана прямо на управляющих схемах из TFT-транзисторов. Дело в том, что использование пассивно-матричных управляющих схем в сочетании с относительно невысокой скоростью работы полимерных "диодов" приводит к неудовлетворительной инерционности экрана. А достоинства активно-матричной технологии были недостижимы из-за неприменимости фотолитографии к тончайшим полимерным пленкам.
Есть все основания полагать, что под боком у ЖК-технологии развивается очень серьезный конкурент. Действительно, технология OLED часто рассматривается экспертами как потенциальная замена не только для ЖК-мониторов, но и плазменных панелей. Дело в том, что OLED-дисплеи имеют целый ряд существенных преимуществ. Они потребляют меньше энергии, не требуют дополнительной подсветки и при этом обеспечивают повышенную яркость, высокую контрастность и частоту регенерации изображения, видимого к тому же под большими углами обзора. Кроме того, OLED-устройства, согласно утверждениям сторонников этой технологии, имеют меньшее время отклика и поэтому лучше приспособлены для быстроменяющегося изображения.
Немаловажным фактором роста популярности OLED-дисплеев может стать и себестоимость массового производства, которая базируется на тонкопленочных и стандартных литографических технологиях. Дело в том, что такая комбинация может обеспечить низкие затраты и высокую надежность всего производственного процесса. Некоторые эксперты полагают, что при условии массового производства стоимость OLED-экранов будет ощутимо ниже, чем у ЖК-панелей. Немаловажной деталью является также тот факт, что такие мониторы работают при напряжении питания в несколько вольт, имеют очень малую массу и толщину. Все это должно сделать технологию привлекательной для производителей электроники и плоскопанельных экранов. Однако до недавнего времени утверждалось, что уровень развития самой технологии еще не позволяет запустить массовое коммерческое производство. Исключения составляют уже устанавливаемые малые экраны в некоторых моделях сотовых телефонов, цифровых камер и ручных компьютеров.
Из недостатков новой технологии стоит отметить относительно низкое "время жизни" (lifetime) излучающих полимеров. Самые большие проблемы возникли с материалами, излучающими синий свет. Сначала они могли работать вообще не дольше тысячи часов, что было явно неприемлемо для практического применения. Достигнутые успехи на сегодняшний день не могут не впечатлять. Хотя в синем спектре перспективные OLED-материалы по-прежнему остаются наименее долговечными, срок их жизни составляет уже около 10 тыс. часов. А осенью прошлого года компании CDT удалось получить OLED-материал с синим свечением, время жизни которого составило 40 тыс. часов.
В рамках данной курсовой работы был исследован нафталин, в частности его электрические свойства. Сложность данного эксперимента заключалась в том, что почти все низкомолекулярные полупроводники, в том числе и нафталин, являются высокоомными проводниками. Поэтому сопротивление измерялось при больших температурах.
Образец нафталина был помещен в кювету из фторопласта. Используя мультиметр ММ-960, было измерено сопротивление органического полупроводника при различной температуре.
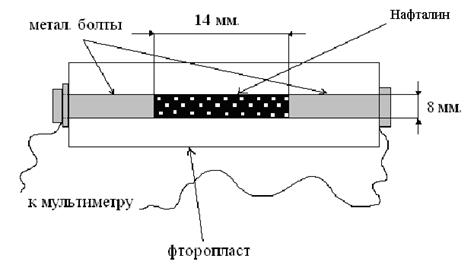
Полученные в ходе эксперимента результаты занесены в таблицу и по ним построен график зависимости логарифма электропроводности ![]() от обратной температуры 1/T.
от обратной температуры 1/T.
Результаты измерений сопротивления при различных температурах.
№ опыта |
Т, температура, К | R, сопротивление, 107 Ом |
1/Т, 104 /К | lnσ |
| 1 | 483 | 1,73 | 48 | -16,8 |
| 2 | 493 | 1,26 | 45 | -15,8 |
| 3 | 503 | 0,91 | 43 | -15,1 |
| 4 | 513 | 0,66 | 42 | -14,7 |
| 5 | 523 | 0,49 | 40 | -14,0 |
| 6 | 533 | 0,37 | 38 | -13,3 |
| 7 | 543 | 0,27 | 37 | -13,0 |
| 8 | 553 | 0,21 | 36 | -12,6 |
| 9 | 563 | 0,16 | 34 | -11,9 |
| 10 | 573 | 0,13 | 33 | -11,6 |
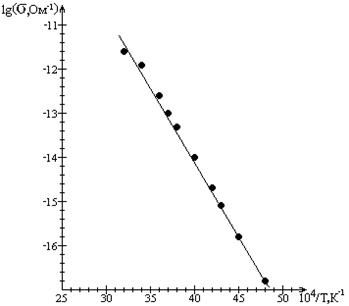
График зависимости логарифма электропроводности ![]() от обратной температуры 1/T.
от обратной температуры 1/T.
Полученная зависимость сходится с теоретическими результатами и соответствует формуле:
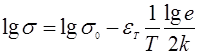 .
.
Используя данную формулу и результаты эксперимента можно вычислить ![]() :
:
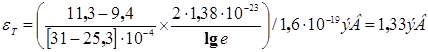 .
.
Вывод:
Полученное значение по порядку совпадает с табличными, но сравнить с точным численным значением невозможно, т.к. значения энергии активации, определяемые для одних и тех же веществ различными авторами, часто оказываются резко различными. Величина ![]() может зависеть от условий приготовления исследуемого образца, от его структуры (аморфный слой, поликристаллический порошок или монокристалл), от условий предварительной его обработки, от того, исследовалась ли электропроводность в вакууме, в атмосфере, в атмосфере газа или на воздухе.
может зависеть от условий приготовления исследуемого образца, от его структуры (аморфный слой, поликристаллический порошок или монокристалл), от условий предварительной его обработки, от того, исследовалась ли электропроводность в вакууме, в атмосфере, в атмосфере газа или на воздухе.
В курсовой работе были рассмотрены особенности класса органических проводников, их практическое применение и экспериментальные разработки в данной области. Хочется сделать вывод, что исследования в области органических проводников являются одним из наиболее перспективных направлений при создании вакуумных фотоэлектронных приборов, спеновых вентилей в высокочувствительной магнитной записи и ячейках памяти персональных компьютеров, гибкой электроники, в частности гибких переносных компьютеров и дисплеев.
Японские ученые, в частности, работают над созданием легких, гибких и, в перспективе, недорогих переносных компьютеров и дисплеев. Владельцы таких компьютеров могли бы по окончании работы свернуть монитор и положить компьютер в сумку (подобные устройства уже представляются компаниями-разработчиками на выставках), а то и в карман. Основными материалами для подобной "гибкой электроники" являются аморфный гидрогенизированный кремний и органические полупроводники.
Таким образом, исследования в области создания новых органических полимеров с полупроводниковыми свойствами являются одним из перспективных направлений в сфере микроэлектроники.
1. Артемьев С.В. , Войкова Е.Д. , Коваль Г.И. , Шевцов М.К. Слои бихромированной желатины, сенсибилизированные для зелено-красной области спектра. / В сб. : Фотохимические процессы регистрации голограмм. Под. Ред. Варачевского В.А. , Л., 1983, с. 131-137
2. Богуславский Л. И., Ванников А. В., Органические полупроводники и биополимеры, М., 1968;
3. Бонч-Бруевич В.Л., Калашников С. Г. Физика полупроводников.- М.: Наука,1977.
4. Вайденбах В.А., Малыгина Г.Г. Ионное равновесие хромовокислой системы. - //Ж. научн. и прикл. фотогр. и кинематогр, 1968, , № 3, с. 165-167.
5. Выговский Ю.Н., Дработурин П.А., Коноп А.Г., Коноп С.П., Малов А.Н. Управление свойствами самопроявляющихся “красных” желатин-глицериновых систем / в. сб.: “Применение лазеров в науке и технике”. В.IX. -ИФ ИЛФ СО РАН: Иркутск -1997 - с. 149-159.
6. Выговский Ю.Н. Фазовые переходы в пленках дихромированного желатина при записи объемных и красных радужных голограмм. Дисс.. к.ф.-м.н., Иркутск: ИГУ.-1997 - 192 с.
7. Гутман Ф., Лайонс Л., Органические полупроводники, пер. с англ., М., 1970
8. Горелик С.С., Дашевский М. Я. Материаловедение полупроводников и металловедение.- М. Металлургия, 1973.
9. Шерстюк В.П. , Кошечко В.Г. , Атаманюк В.Ю. Исследование кинетики и механизма окисления трифениламинов хроматом натрия. //Журнал общей химии, 1980, 50, № 10, с. 2153-2159.
10. Шерстюк В.П. , Дилунг И.И. , Мазур Л.В. , Лялецкая О.А. Фотохимические и темновые реакции соединений хрома (VI), механизм дубления и фотографические свойства хромированных коллоидов. / В сб. “III Всесоюзн. Конф. по бессереб. и необ. процессам”. Фотохим. методы регист. информации и полупроводниковая фотограф.”, Вильнюс, май 1980, с. 205.
Похожие рефераты:
Химия, элементы таблицы Менделеева
Лекции по твердотельной электронике
Программа для поступающих в вузы (ответы)
Концепции современного естествознания
Основные качества полупроводников
Перспективы развития экологического сознания школьников при изучении темы "Полимеры" в курсе химии
Лекции - преподаватель Григорьев Владимир Калистратович
Разработка анализатора газов на базе газового сенсора RS 286-620
Разработка школьного элективного курса "Полимеры вокруг нас"