| Скачать .docx |
Дипломная работа: Поиск новых фторидофосфатов лития и переходных металлов
Министерство образования Российской федерации
Государственное образовательное учреждение высшего профессионального образования
«РОСТОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ»
Химический факультет
ДИПЛОМНАЯ РАБОТА
на тему: «ПОИСК НОВЫХ ФТОРИДОФОСФАТОВ ЛИТИЯ И ПЕРЕХОДНЫХ МЕТАЛЛОВ»
Студент 6 курса
Ворона М.Л.
Научный руководитель,
доцент, кандидат хим. наук
Налбандян В. Б.
Рецензент
Ст. научный сотр. НИИФОХ, кандидат хим. наук
Медведева Л. И.
Нормоконтролер
Богатырева Н.К.
г. Ростов-на-Дону
2004
Содержание
Введение
1. Литературный обзор
1.1. Литий-ионные аккумуляторы
1.2. Смешанные фосфаты лития и переходных металлов
1.3. Смешанные фторидофосфаты щелочных и переходных металлов.
2. Исходные вещества и методы эксперимента
2.1. Исходные вещества и их анализ
2.2. Проведение синтезов
2.3. Рентгенофазовый анализ
3. Результаты и их обсуждение
3.1. Фторидофосфат никеля-лития
3.2. Фторидофосфат кобальта-лития
3.3. Фторидофосфаты железа-лития и марганца-лития
3.4. Опыт по окислению
4. Выводы и перспективы
Список использованных источников
Введение
Для современной техники очень важны энергоемкие и портативные аккумуляторы. Используемые в них электродные материалы обладают рядом недостатков и поэтому актуален поиск новых материалов. В частности, для положительного электрода литий-ионного аккумулятора нужны материалы, способные к быстрому и обратимому внедрению–извлечению лития с достаточно высоким потенциалом относительно лития. Данная работа посвящена поиску новых смешанных фторидофосфатов лития с переходными металлами, легко меняющими степень окисления: марганцем, железом, кобальтом, - и исследованию возможности окислительного извлечения лития из них, как первому шагу к испытанию их в качестве электродных материалов.
1. Литературный обзор
1.1. Литий – ионные аккумуляторы
Становление технологий никель-металлгидридных и литий-ионнных аккумуляторов вытесняет известные никель-кадмиевые аккумуляторы.
Батарея - устройство для накопления энергии, или, когда речь идёт о современных технологиях, под батареей обычно понимают автономную химическую систему, производящую электроэнергию [1]. Батарея составлена из нескольких электрохимических ячеек, соединенных последовательно или параллельно, чтобы обеспечить необходимое напряжение и емкость. Каждая ячейка состоят из положительного и отрицательного электрода, отделенных жидким или твердым электролитом. Побуждением для использования технологии батареи, основанной на литии, как аноде, полагают тот факт, что этот металл является очень сильным восстановителем и очень легким, тем самым обеспечивается высокая энергоемкость.В зависимости от типа положительного электрода, литиевые элементы могут создавать напряжение от полутора до 4 вольт, что выше, чем у любых других элементов. Поскольку литий взаимодействует с водой и спиртами, в качестве электролитов используются апротонные жидкости – растворы гексафторфосфата или гексафторарсената лития в смеси эфиров (диметилкарбоната, этиленкарбоната, пропиленкарбоната, диметоксиэтана и т.д.).
Таким образом, достигается превращение накопленной химической энергии в энергию электрическую (разрядка). Как только к электродам присоединят внешний источник тока, т.е. пропустят через них электрический ток, тогда электрическая энергия в них преобразуется в химическую (перезарядка). Ячейки характеризуются количеством запасаемой электрической энергии или заряда в расчете на единицу массы (Вт*час/кг или A*час/кг) или единицу объема (Вт*час/л или А*час/л), разрядным напряжением и циклируемостью. Литий-ионные батареи из-за их высокой энергетической плотности и гибкости конструкции в настоящее время превосходят другие системы, составляя 63 % оцениваемого всемирного рынка переносных батарей [2].
Первоначально в качестве материала отрицательного полюса использовался чистый литий. Но, как выяснилось, при первом контакте литий восстанавливает раствор, и на его поверхности образуется пленка из продуктов восстановления. Пленка эта достаточно тонкая (несколько нанометров), сплошная и проводящая, причем носителями заряда будут ионы лития (по сути дела, получается твердый электролит). Пленка становится барьером, и дальше металлический литий с электролитом не взаимодействует.Таким образом, на аноде литиевого элемента в аккумуляторе при разряде будет протекать реакция: Li ® Li+ + e. А вот при заряде или так называемом катодном осаждении лития, происходит перемещение ионов лития с положительного электрода и осаждение на отрицательном электроде. Этот процесс, во-первых, может вызвать рост литиевых дендритов и короткое замыкание — верный путь к возгоранию или взрыву элемента, а во-вторых, при осаждении лития образуется свежая очень активная поверхность, мгновенно реагирующая с электролитом.На этой поверхности сразу нарастает пленка, предотвращая электрический контакт с самим электродом.Из-за этого в аккумуляторы с металлическим литиевым электродом приходится закладывать избыточное количество лития, с расчетом на то, что часть его потеряется. Вот поэтому литиевые аккумуляторы (с электродом из чистого металлического лития) теряют свое значение. На смену им пришли так называемые литий-ионные аккумуляторы, где отрицательным электродом служит не чистый литий, а фаза внедрения лития в подходящую матрицу с достаточно низким электродным потенциалом.
Углерод оказался очень удобной матрицей для помещения в него лития. Удельный объем углеродных материалов при этом изменяется не сильно — даже при внедрении достаточно большого количества лития он увеличивается не более чем на 10%. Чем больше лития внедрено в углерод, тем отрицательнее потенциал электрода [3].
Такие элементы работают без подзарядки в полтора раза дольше никель-металлгидридных. Кроме того, в литий-ионных элементах не наблюдаются эффекты памяти, которыми славились ранние никель-кадмиевые элементы. С другой стороны, внутреннее сопротивление у современных литиевых элементов выше, чем у никель-кадмиевых. Соответственно, они не могут обеспечить больших токов. Литий-ионная батарея выдерживает многократные подзарядки: 500-1000 циклов [4].
В качестве положительного электрода используются соединения переходных металлов, способных к легкому изменению степени окисления с обратимым внедрением-извлечением лития. Чтобы ячейка могла разряжаться большими токами, нужна большая электронная проводимость и высокий коэффициент диффузии лития, а для этого в структуре должны быть каналы для миграции лития. А чтобы разряд и заряд были обратимыми, структура должна быть достаточно жесткой, чтобы сохраняться практически неизменной и в отсутствие лития.
Наиболее широко распространенным материалом является кобальтит лития LiCoO2 . При заряде ионы лития извлекаются из кобальтита и внедряются в углерод:
(положительный электрод) LiCoO2 ® x Li+ + x e + Li1-x CoO2
(отрицательный электрод) Li+ + e + 6 C ® LiC6
При разряде идут обратные процессы, степень окисления кобальта при этом снижается [3].
Сложный оксид LiCoO2 обладает слоистой структурой, в которой ионы лития и кобальта упорядочены в чередующихся плоскостях. Наличие плоскостей, занятых исключительно ионами лития, обеспечивает возможность почти полного извлечения щелочного металла и тем самым применимость данного соединения в качестве катодного материала в химических источниках тока. Но продукт полного извлечения – слоистый CoO2 – очень неустойчив, и на практике циклирование ведут в диапазоне x от 0 до приблизительно 0,5[2].
При десяти циклах заряд (4,2 В) - разряд (3,5 В) начальная удельная разрядная емкость 145 A*час/кг. Потери разрядной удельной емкости 0,1% на 1 цикл [5].
К недостаткам кобальтита лития относят то, что при многократном циклировании часть ионов кобальта перемещается в литевые слои, слоистая структура перестраивается в каркасную типа шпинели, и движение ионов лития затрудняется, а также высокую стоимость и токсичность [2].
Поэтому ведутся интенсивные поиски и исследования альтернативных материалов. В частности, большое число работ посвящено легированию LiCoO2 , структурно родственным ему соединениям LiMnO2 , LiNiO2 , фазам типа шпинели на основе LiMn2 O4 и др. В частности, хорошо зарекомендовали себя фазы типа оливина LiMPO4 (где M = Mn, Fe, Co, Ni), описываемые ниже.
1.2. Смешанные фосфаты лития и переходных металлов
Двойные фосфаты, имеющие общую формулу LiMPO4 (где M = Mn, Fe, Co, Ni), изоструктурны оливину - силикату магния и железа (Mg,Fe)2 SiO4 .
Таблица 1
Параметры решетки и разрядные характеристики соединений LiMPO4 [6- 9]
| M | a, Е | b, Е | c, Е | U, В | Емкость, А*час/кг |
| Mn | 10,45 | 6,11 | 4,75 | 4,1 | 140 |
| Fe | 10,31 | 6,00 | 4,69 | 4,3 | 148 |
| Co | 10,20 | 5,92 | 4,68 | 4,8 | 86 |
| Ni | 10,20 | 5,92 | 4,68 |
Фосфаты LiMPO4, где M = Mn, Co, Ni получены в ходе взаимодействия карбоната лития, оксида металла (MO или MnO2 ) и дигидрофосфата аммония - (NH4 )2 HPO4 при температуре 350 °C, которую затем повышали до 780 °C и выдерживали 18 часов на воздухе [6]. LiFePO4 получен аналогично, но в инертной атмосфере [10].
1.3. Смешанные фторидофосфаты щелочных и переходных металлов
Просмотр реферативных журналов, баз данных PDF-2 и ICSD обнаружил только три фазы формульного типа A+ 2 MPO4 F, из них с литием только одна: Li2 NiPO4 F [11]. Известны также Na2 MnPO4 F [12], Na2 MgPO4 F [13], Na4,6 FeP2 O8,6 F0,4 [14, 15, 16, 17].
Структура Li2 NiPO4 F(рис. 1) определена рентгенографически на монокристалле [11]. Она относится к ромбической сингонии (пространственная группа Pnma, параметры a= 10.473(3) Е, b= 6.2887(8) Е, c= 10.846(1) Е, Z=8). В структуре можно выделить рутиловые цепи из октаэдров NiO4 F2 , соединенных ребрами, вытянутые вдоль оси y. Эти цепи соединены в двух остальных измерениях тетраэдрами PO4 . В пустотах каркаса размещаются катионы лития. Половина их находится в уплощенных тетраэдрах из четырех атомов кислорода, четверть – в квадратных пирамидах из 4 O + 1 F и еще одна четверть в сильно асимметричной координации, где трудно сделать однозначный выбор между КЧ 4,5,6. Достаточно короткие (до 3,21 Е) расстояния Li-Li соединяют все позиции лития в двумерную сеть в плоскости y0z (рис. 2). Это позволяет ожидать достаточно высокую подвижность ионов лития в каркасе и возможность их извлечения с окислением никеля и сохранением исходного каркаса:
Li2 Ni2+ PO4 F ® LiNi3+ PO4 F + Li+ + e ® Ni4+ PO4 F + 2 Li+ + 2 e
Но сведений о таких свойствах Li2 NiPO4 F в литературе не обнаружено. Можно было бы ожидать существование аналогичных фаз, содержащих на месте никеля другие катионы близкого размера с переменной степенью окисления (табл. 2), но никаких сведений о них в литературе также не обнаружено.
Таблица 2
Эффективные кристаллохимические радиусы [18] некоторых двухзарядных катионов в октаэдрической координации в высокоспиновом состоянии
| M | Mn2+ | Fe2+ | Co2+ | Ni2+ |
| VI R, Е | 0,97 | 0,92 | 0,885 | 0,83 |
В данной работе поставлена задача получения новых фаз состава Li2 MPO4 F, где M = Mn, Fe, Co, и исследования возможности окислительного извлечения лития из них и из ранее известного никелевого соединения. Предполагалось, что за счет удвоенного содержания лития можно будет повысить емкость электродного материала по сравнению с фазами типа оливина (табл. 3).
Таблица 3
Теоретические удельные емкости некоторых известных и предполагаемых материалов положительного электрода литий-ионного аккумулятора
| Восстановленная форма | Окисленная форма | Емкость, А*час/кг |
| LiMO2 (M = Co, Ni) | Li0.5 MO2 | 140 |
| LiMPO4 (M = Mn, Fe, Co, Ni) | MPO4 | 170 |
| Li2 MPO4 F (M = Mn, Fe, Co, Ni) | LiMPO4 F | 144 |
| MPO4 F | 288 |
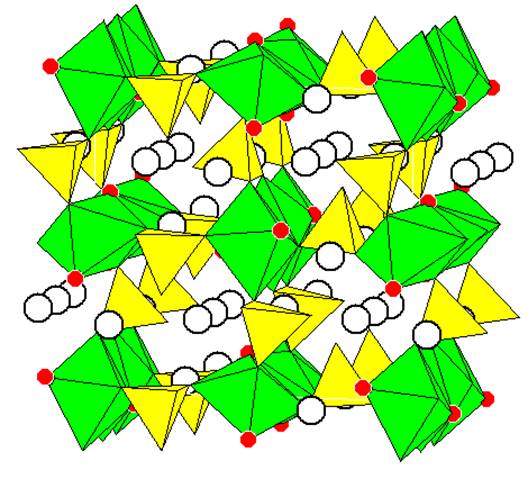 |
Рисунок 1
Полиэдрическое изображение кристаллической структуры Li2 NiPO4 F [10]
Зеленым цветом показаны октаэдры вокруг катионов никеля, желтым – тетраэдры PO4, красным – ионы фтора (в остальных вершинах – кислород), светлыми кружками показаны ионы лития.
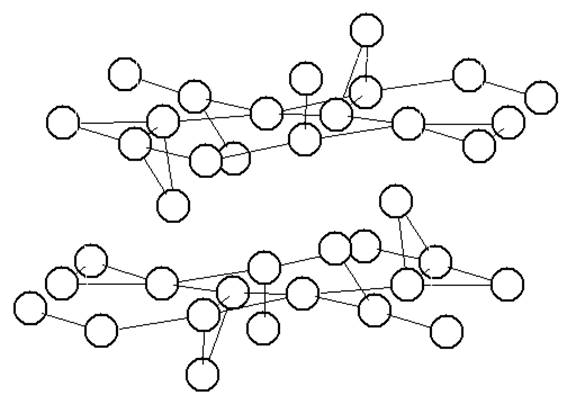 |
Рисунок 2
Система позиций лития в структуре Li2 NiPO4 F. Соединены позиции, отстоящие друг от друга не более чем на 3,21 Е.
2. Исходные вещества и методы экспериментов
2.1. Исходные вещества и их анализ
Фосфор, фтор и литий вводили в виде дигидрофосфата аммония, высушенного при 100 °С, фторида и карбоната лития, высушенных при 200 °С. Реактивный оксид никеля (серый, нестехиометрический) прокаливали при 900 °C для превращения в зеленый стехиометрический NiO. Реактивный оксид кобальта (+2) использовали в непрокаленном виде (рентгенофазовым анализом проверено, что это действительно CoO, а не Co3 O4 ). Для введения переходных металлов испытаны и другие реагенты: карбонаты кобальта и марганца, ацетат никеля, а также оксалаты марганца и железа (+2), осажденные из водных растворов. Для проведения данной части экспериментов брали растворимые соли: сульфат железа (+2) и хлорид марганца (+2), растворяли их в горячей дистиллированной воде и приливали к ним горячий раствор оксалата аммония. После охлаждения осадки отфильтровывали на воронке Бюхнера, промывали дистиллированной водой до удаления сульфат- или хлорид-ионов и высушивали при комнатной температуре несколько дней.
Нет уверенности в том, что эти карбонаты, оксалаты и ацетат точно соответствуют идеальным формулам: при хранении возможны потеря или приобретение воды, гидролиз, окисление. Поэтому потребовалось провести их анализ. Для этого по три параллельных пробы каждого из исходных веществ прокаливали до постоянной массы и взвешивали в виде оксидов. Температуру прокаливания выбирали на основе литературных данных о стабильности весовых форм: для получения Fe2 O3 , NiO – 900 °С, для получения Co3 O4 и Mn2 O3 - 750 °С [19, 20, 21].
2.2. Проведение синтезов
При нагревании фторида лития с дигидрофосфатом аммония возможно улетучивание фтороводорода. Поэтому проведение синтеза в одну стадию вряд ли возможно. Сначала нужно получить LiMPO4 , и лишь после полного удаления воды можно добавлять фторид лития.
Таким образом, можно выделить две стадии.
(1) 2NH4 H2 PO4 +Li2 CO3 + 2MO ® 2 LiMPO4 + 2NH3 + CO2 + 2H2 O.
Здесь MO – это либо оксид (NiO, CoO), либо соединение, разлагающееся до оксида.
(2) LiMPO4 + LiF ® Li2 MPO4 F
Навески веществ смешивали и растирали в яшмовой ступке до полной однородной массы, затем прессовали таблетки, выдерживали при температуре 150-170 °C 2 часа для удаления большей части летучих компонентов (если сразу нагреть до более высоких температур, то происходит оплавление и однородность таблетки нарушается). Затем температуру постепенно повышали, периодически перетирая смесь, до получения практически чистых LiMPO4 . Обжиги проводили либо в муфельной печи, либо в инертной атмосфере в трубчатой печи.
Ввиду отсутствия инертных газов в баллонах, пришлось получать азот нагреванием водного раствора хлорида аммония и нитрита бария. Колба, в которой происходила основная реакция по получению азота (экзотермическая реакция, небольшое нагревание), соединялась с двумя промывалками с сернокислым раствором бихромата калия для улавливания возможных примесей аммиака и оксида азота, далее шла накаливаемая трубка с пористыми медными гранулами для очистки от кислорода и оксидов азота, потом с силикагелем для грубой осушки и две промывалки с концентрированной серной кислотой для более полного улавливания водяных паров. Эти промывалки соединялись с трубкой, в которой находились смеси веществ в спрессованном виде в никелевых лодочках. Вначале через установку пропускали трехкратный объем азота для удаления воздуха и лишь потом начинали нагревание. После завершения обжига образцы охлаждали в токе азота, дабы не допустить окисления воздухом.
Продукты проверяли рентгенофазовым анализом и переходили ко второй стадии экспериментов, для этого полученные таблетки перетирали с рассчитанной навеской фторида лития и, спрессовав, продолжали обжиг либо в муфельной печи, либо в инертной атмосфере в трубчатой печи по уже рассмотренной технологии. Чтобы обеспечить более полное связывание фосфата, фторид лития вводили в пятипроцентном избытке. Этот избыток составляет лишь 0,7 масс. % смеси и менее существенен, чем примесь не прореагировавшего фосфата.
2.3. Рентгенография
Рентгенофазовый анализ производился на дифрактометре ДРОН – 2.0 в медном Кa - излучении. Данное излучение не очень подходит для соединений, в которых присутствуют железо и особенно кобальт, так как оно сильно поглощается атомами этих элементов и возбуждает их собственное рентгеновское излучение. В результате дифракционные максимумы ослабляются, и резко возрастает фон. Поэтому снижается чувствительность фазового анализа, уменьшается число наблюдаемых отражений и ухудшается точность их измерения из-за сильных флуктуаций интенсивности. Чтобы преодолеть эти затруднения, следовало бы использовать рентгеновскую трубку с другим анодом, например, кобальтовым (но тогда бы возникли те же проблемы с соединениями марганца) или установить монохроматор на дифрагированном пучке. Но у нас не было такой возможности, поэтому для уменьшения статистических ошибок съемку кобальтового соединения приходилось повторять по несколько раз.
При фазовом анализе применялась база порошковых дифракционных данных PDF-2.
3. Результаты и их обсуждение
3.1. Фторидофосфат никеля-лития
Синтез проводился в две стадии, как описано выше. Если исходным веществом был ацетат никеля, то при его разложении происходило частичное восстановление никеля (образец чернел), поэтому требовался обжиг в окислительной атмосфере. Если же исходное вещество - оксид никеля, то и первый, и второй обжиги можно проводить и на воздухе, и в азоте, результаты практически одинаковые. На первой стадии при температуре последнего обжига 750°С получен почти чистый желтый LiNiPO4 с небольшой примесью NiO, а после 680°С содержание примесей было несколько больше, и образец был серого цвета. Но в обоих случаях на второй стадии - при обжиге с LiF (750 °С, 2-4 часа) - получен практически чистый Li2 NiPO4 F серо-зеленого цвета. В имеющейся базе порошковых дифракционных данных нет его рентгенограммы, но она была рассчитана на основе структурных данных [11] с помощью программы Lazy Pulverix, и экспериментальные данные хорошо совпали с расчетными.
3.2 Фторидофосфат кобальта-лития
В согласии с литературными данными [6], LiCoPO4 удалось получить на воздухе. На первой стадии смесь исходных соединений поместили в сушильный шкаф при температуре 170 °C, выдержали 2 часа, затем переместили в муфельную печь, и медленно нагрели до 680 °C, выдержав 40 минут, после чего тщательно растерли и выдержали при 750°C 30 минут. Получен порошок фиолетового цвета, по данным рентгенофазового анализа соответствующий фосфату кобальта-лития.
Однако на второй стадии, после его реакции с LiF при 750°С, вместо ожидаемого Li2 CoPO4 F обнаружено большое количество Co3 O4 в смеси с исходным LiCoPO4 и неизвестными фазами. Поскольку без фторида лития этого оксида кобальта не наблюдалось, можно предположить, что к его образованию привело сочетание сразу нескольких побочных явлений: гидролиз фторида водяным паром увеличил содержание Li2 O, поэтому менее основный CoO был вытеснен из фосфата, чему способствовало его окисление до Co3 O4 . Поэтому присутствие кислорода воздуха и водяных паров мешает при твердофазном синтезе фторидофосфата кобальта.
После этого весь эксперимент последовательно проведен в инертной атмосфере. Для чего на первой стадии снова приготовили смесь веществ, спрессовав, поместили в трубчатую печь выдержали в интервале температур от 120 до 300 °C около часа, затем стали повышать температуру на 50°C каждые 10-15 минут, доведя до 750 °C, выдержали 1,5 часа. Преимуществом был тот факт, что реакция проходила при постоянном токе азота, после охладили систему в азоте, извлекли таблетку и растерли ее, порошок фиолетового цвета. Образец, взятый на рентгенофазовый анализ, показал наличие фосфата кобальта-лития и незначительного количества примесей по сравнению с тем порошком, который был получен в воздухе. Затем добавили расчетное количество LiF и, спрессовав таблетку, поместили в трубчатую печь, нагрели в токе азота до 750 °C, выдержали 2 часа , затем охладили систему в присутствии азота, таблетку извлекли и растерли, полученный темно-фиолетовый порошок проверили с помощью рентгенофазового анализа.
На рентгенограмме отсутствовали пики исходных LiCoPO4 , LiF, оксидов кобальта. По расположению и интенсивности пиков рентгенограмма этого продукта оказалась сходна с расчетной рентгенограммой Li2 NiPO4 F, что позволило полностью проиндицировать ее на основе аналогичной ромбической элементарной ячейки (табл. 4). Впрочем, попытка механического переноса индексов hkl с одной рентгенограммы на другую первоначально не привела к удовлетворительному результату. Лишь после нескольких проб и ошибок выяснилось, что замещение никеля кобальтом ведет к анизотропному изменению параметров (a уменьшается, b , c и объем возрастают, см. табл. 5), поэтому некоторые линии на рентгенограмме меняются местами.
Правильность индицирования подтверждается хорошим согласием вычисленных и измеренных значений углов (табл. 4). Найденный объем ячейки, несколько больший, чем у никелевого аналога (табл. 5), хорошо согласуется с соотношением размеров ионов никеля и кобальта (табл. 2). Таким образом, синтезировано новое соединение Li2 CoPO4 F, изоструктурное Li2 NiPO4 F.
Таблица 4
Результаты индицирования рентгенограммы нового соединения Li2 CoPO4 F в сравнении с рентгенограммой Li2 NiPO4 F, рассчитанной на основе его кристаллической структуры с помощью программы Lazy Pulverix. Параметры решетки уточнены с помощью программы Celref 3 и приведены в таблице 5.
| hkl | Li2 NiPO4 F | Li2 CoPO4 F | ||||
| I | 2qвыч | Iэкс | 2qэкс | 2qвыч | D (2q) | |
| 002 | 86 | 16.34 | 60 | 16.33 | 16.32 | 0.01 |
| 200 | 100 | 16.93 | 85 | 17.03 | 16.98 | 0.05 |
| 211 | 40 | 23.58 | 50 | 23.50 | 23.51 | - 0.01 |
| 013 | 32 | 28.48 | 25 | 28.40 | 28.35 | 0.05 |
| 311 | 14 | 30.42 | 20 | 30.44 | 30.42 | 0.02 |
| 022 | 45 | 32.93 | 40 | 32.59 | 32.59 | 0.00 |
| 004 | 36 | 33.03 | 40 | 32.97 | 32.98 | - 0.01 |
| 400 | 47 | 34.25 | 100 | 34.31 | 34.36 | - 0.05 |
| 222 | 45 | 37.25 | 50 | 36.98 | 36.98 | 0.00 |
| 410 | 7 | 37.20 | 10 | 37.25 | 37.23 | 0.02 |
| 402 | 5 | 38.17 | 20 | 38.25 | 38.26 | - 0.01 |
| 123 | 16 | 38.92 | 10 | 38.62 | 38.62 | 0.00 |
| 214 | 11 | 40.10 | 25 | 40.00 | 40.01 | - 0.01 |
| 224 | 16 | 47.56 | 30 | 47.29 | 47.31 | - 0.02 |
| 422 | 32 | 48.24 | 20 | 48.08 | 48.08 | 0.00 |
| 424 | 15 | 56.99 | 25 | 56.82 | 56.83 | - 0.01 |
| 026 | 28 | 58.93 | 10 | 58.65 | 58.65 | 0.00 |
Таблица 5
Сравнение параметров ромбических решеток Li2 MPO4 F (в скобках – стандартное отклонение последней значащей цифры)
| M | a Е | b Е | c Е | V |
| Ni | 10.473(3) | 6.2887(8) | 10.846(1) | 714.3 |
| Co | 10.440(5) | 6.368(9) | 10.863(8) | 722.3(8) |
3.3. Соединения, содержащие марганец и железо
Попытки синтеза Li2 FePO4 F проводили в инертной атмосфере, так как соединения железа (2+) быстро окисляются на воздухе. По той же причине трудно подобрать устойчивую весовую форму исходного соединения железа (2+). В данной работе для приготовления промежуточного соединения LiFePO4 использовали FeC2 O4 *2H2 O, желтый осадок которого был получен и проанализирован, как описано выше. В литературе имеются противоречивые сведения о продуктах разложения чистого оксалата железа. По одним данным, получается оксид железа (2+), по другим - пирофорный металл. Мы предполагали (как и подтвердилось впоследствии), что для окисления этого металла будет достаточно примеси кислорода в азоте. Если бы при первом опыте был обнаружен металл, то можно было бы в дальнейшем использовать сочетание FeC2 O4 *2H2 O + Fe2 O3 для получения заданной степени окисления железа.
Смесь оксалата железа, карбоната лития и дигидрофосфата аммония, спрессовав, поместили в трубчатую печь и при постоянном токе азота выдержали в интервале температур от 120 до 300 °C около часа, затем стали повышать температуру на 50°C каждые 10-15 минут, доведя до 750 °C, выдержали 1,5 часа , после чего охладили систему в азоте, извлекли таблетку и растерли ее. Получен порошок черного цвета, притягивающийся к магниту. Но по данным рентгенофазового анализа ферромагнитная фаза - это не металлическое железо, а магнетит Fe3 O4 . Вторую стадию, реакцию с LiF, проводили при 750 °C в течение 2 часов в токе азота. В результате таблетка сильно деформировалась (что указывает на появление небольшого количества жидкой фазы), а рентгенофазовый анализ показал смесь LiFePO4 + LiF. Таким образом, ожидаемое соединение Li2 FePO4 F не получилось.
Согласно литературным данным [6], LiMnPO4 может быть синтезирован на воздухе при 780°С. Поскольку соединения марганца (2+) окисляются почти так же легко, как соединения железа (2+), это казалось маловероятным и в данной работе не подтвердилось. После обжига на воздухе на рентгенограммах неизменно присутствовали яркие отражения Mn2 O3 . Поэтому синтез был проведен по той же схеме, что и в случае железа - через оксалат марганца (2+) в азоте. При температуре заключительного обжига 750°С в течение 1,5 часа получен практически чистый LiMnPO4 серого цвета. Но взаимодействия LiF с LiMnPO4 не обнаружено даже вблизи температуры плавления смеси.
Отсутствие в этих опытах соединений Li2 MPO4 F (M = Fe, Mn) нельзя объяснить ни окислением (поскольку найденные фазы соответствуют желаемой степени окисления железа и марганца), ни гидролизом фторида (фторид лития обнаружен), ни кинетическими затруднениями (температура была достаточно высокой, близкой к плавлению, и соединения никеля и кобальта в тех же условиях получались легко). Очевидно, соединения Li2 MPO4 F (M = Fe, Mn) в рассматриваемых условиях термодинамически менее стабильны, чем смеси LiMPO4 + LiF. Вероятно, катионы железа и марганца чрезмерно крупные (см. табл. 2) для стабильности данного типа структуры).
Было бы интересно проверить влияние давления на направление реакции LiMPO4 + LiF = Li2 MPO4 F. Для этого по известным параметрам решетки рассчитаны формульные объемы реагентов и продуктов (табл. 6). Из нее видно, что реакция идет с небольшим увеличением объема, поэтому высокие давления будут, вероятно, смешать равновесие влево, то есть еще больше дестабилизировать фторидофосфаты.
Таблица 6
Сравнение объемов (в кубических ангстремах) в расчете на формульную единицу реагентов и продуктов
| M | V/Z | Δ V | ||
| LiF | LiMPO4 | Li2 MPO4 F | ||
| Ni | 16,35 [22] | 68,65-69,24 [6, 23, 24] | 89,29 [11] | 3,7-4,3 |
| Co | 16,35 | 70,80-71,03 [25, 26] | 90,30 | 2,9-3,1 |
3.4. Опыт по окислению
Суть этого опыта сводится к попытке окисления полученного фторидофосфата кобальта-лития раствором брома в метаноле с целью извлечения части или всего лития с сохранением каркаса MPO4 F.
Li2 CoPO4 F + 1/2Br2 ® LiCo+3 PO4 F + LiBr
Li2 CoPO4 F + Br2 ® Co+4 PO4 F + 2 LiBr
Метанол был выбран потому, что он, в отличие от неполярных жидкостей, растворяет не только бром, но и бромид лития, и в то же время, в отличие от воды, не образует с бромом кислот, которые могли бы реагировать с нашим фосфатом. Метанол предварительно осушали кипячением с оксидом кальция и перегоняли.
Бюкс с навеской фторидофосфата кобальта-лития поместили в бокс, туда же поместили силикагель для поглощения паров воды и брома, метанол и ампулу с бромом. Все операции по бромированию проводили в боксе (бром – яд, очень хорошо улетучивается), для этого осторожно вскрыли ампулу с бромом и вылили ее содержимое в заранее подготовленную колбу с метанолом, перемешали. В расчете 15 г брома на 100 мл раствора, что соответствует 0,94 моль/л.
После чего прилили приблизительно трехкратный избыток раствора брома в метаноле в бюкс с фторидофосфатом кобальта-лития и оставили на неделю в боксе для процесса окисления, периодически встряхивая. Затем раствор декантировали, залили свежую порцию раствора и обработку повторили в течение еще олной недели. По истечении данного срока слили раствор и промыли осадок метанолом методом декантации.
Продукт высушили в вакуум-эксикаторе, отобрали пробы и проанализировали их на степень окисления кобальта.
Для этого к ним прилили по 20 мл 0,1 М раствора FeSO4 в 1 M H2 SO4 и нагрели для растворения осадка. Параллельно проводили холостые опыты с 20 мл того же раствора, но без анализируемого вещества.
При титровании перманганатом обнаружилась полная сходимость холостых опытов и опытов, в которых вместе с восстановителем содержались исследуемые соединения. Таким образом, никакого окисления фторидофосфата лития-кобальта не обнаружено. По данным рентгенофазового анализа изменений тоже не наблюдается.
Напрашивается вывод, что данное соединение может окисляться более сильным окислителем и нуждается в более детальном рассмотрении, выходящем за рамки дипломной работы. Если у него потенциал относительно лития около 4 В, то бром, очевидно, недостаточно сильный окислитель для извлечения лития
4. Выводы и перспективы
В результате работы получено одно новое соединение состава Li2 CoPO4 F, показана его изоструктурность с никелевым аналогом. Установлено отсутствие таких соединений с железом и марганцем на месте никеля. Новое соединение может представить интерес как материал положительного электрода литий-ионного аккумулятора, но для этого нужно провести его электрохимические испытания, что не входило в задачи данной работы.
Список использованных источников
1. Элементы питания. Прошлое, будущее и настоящее. http://www.fotolux.com.ua/article/anatomi_13.htm
2. Tarascon J.-M., Armand M./Issues and challenges facing rechargeable lithium batteries// Nature. 2001. V. 414. P. 359-367.
3. Скундин А.М./ Меньше, чем маленький…// Химия и жизнь, 2003, № 7-8.
4. Элементы питания. Прошлое, будущее и настоящее. http://www.fotolux.com.ua/article/anatomi_20.htm.
5. Литий кобальтит. Информация о разработках компании «Балтийская мануфактура». http://www.soli.ru/new_study.htm.
6. Krabbenhoft D., McCarthy G.// ICDD Grant-in-Aid. 1980. (Цит. по PDF-2, № 32-552, 32-578, 33-804).
7. Guohua Li, Hideto Azuma, Masayuki Tohda/ LiMnPO4 as the Cathode for Lithium Batteries// Electrochemical and Solid-State Letters, 2002, V. 5, Iss 6, pp. A135-A137 (Цит. по реферату из Интернет).
8. Yang S., Song Y., Zavalij P. Y., Whittingham M. S.// Reactivity, stability and electrochemical behavior of lithium iron phosphates./ Electrochemistry Communications, 2002, 4, P. 239–244. (Цит. по реферату из Интернет).
9. Amine K., Yasuda H., Yamachib M.// Olivine LiCoPO4 as 4.8 V Electrode Material for Lithium Batteries/ Electrochemical and Solid-State Letters. 2000. 3 (4). P. 178-179. (Цит. порефератуизИнтернет)
10. S. Yang, P.Y. Zavalij, M.S. Whittingham.// Electrochemistry Communications/ 2001. 3. P.505. (Цит. по реферату из Интернет)
11. Dutreilh M., Chevalier C., El-Ghozzi M., Avignant D.// Synthesis and crystal structure of a new lithium nickel fluorophospate Li2 (NiF(PO4 )) with an ordered mixed anionic framework/ Journal of Solid State Chemistry. 1999. V. 142. P.1-5. (Цит. по ICSD, № 50588).
12. Yakubovich O.V., Karimova J.V., Mel`nikov O.K./ The mixed anionic framework in the structure of Na2 (MnF(PO4 ).// Acta Cryst. C. 1997. V. 53. P. 395-397. (Цит. по ICSD)
13. Sean H. Swafford and Elizabeth M. Holt/ New synthetic approaches to monophosphate fluoride ceramics: synthesis and structural characterization of Na2 Mg(PO4 )F and Sr5 (PO4 )3 F.// Solid State Sciences. 2002. V. 4. P. 807-812.
14. Расцветаева Р.К., Максимов Б.А., Тимофеева В.А.// Кристаллическая структура нового Na,Fe-фосфата Na5 Fe(PO4 )F2 ./ ДАН СССР. 1996. Т. 350. № 4. С. 499-502.
15. Голубев А.М., Максимов Б.А., Клокова Н.Е, Мельников О.К., Тимофеева В.А., Сорокин Н.И., Симонов В.И.// Кристаллическая структура натрий-железо (III) – фторофосфата Na4,6 FeP2 O8,6 F0,4 ./ Кристаллография. 1989. Т. 34. Вып. 6. С. 1574.
16. Максимов Б.А., Клокова Н.Е, Радаев С.Ф., Симонов В.И.// Уточнение атомной структуры ионного проводника Na4+x FeP2 O8+x F1-x ./ Кристаллография. 1992. Т. 37. Вып. 5. С. 1143-1151.
17. Максимов Б.А., Тамазян Р.А., Клокова Н.Е, Петржичек В., Попов А.Н., Симонов В.И.// Несоизмерная модуляция в структуре Na9 {Fe2 [PO4 ]4 F2 } при 623 К./ Кристаллография. 1992. Т. 37. Вып. 5. С. 1152-1163.
18. Shannon R.D.// Acta Crystallogr. 1976. V. A32. № 5. Р. 751.
19. Савостина В.М., Пешкова В.М.// Аналитическая химия никеля, М.: «Наука». 1966.
20. Пятницкий И.В.// Аналитическая химия кобальта, М.: «Наука». 1965.
21. Лаврухина А.К., Юнина Л.В.// Аналитическая химия марганца, М.: «Наука». 1974.
22. Van Velthuizen, J., Chao G.// Can. Mineral.1989. 27. P.125. (Цит. по PDF-2, № 45-1460).
23. Abrahams J., Easson K.S.// Structure of lithium nickel phosphate/ Acta Crystallographica. 1993. 49. P.925-926. . (Цит. по реферату)
24. Warda S.A., Lee S-L.// Refinement of the crystal structure of lithium nickel phosphate, LiNiPO4 ./ Zeitschrift fuer Krystallographie – New Crystal Structures. ZKNSF 212. 1997. P. 319. (Цит. по реферату)
25. Kubel F.// Crystal Structure of lithium cobalt double orthophosphate, LiCoPO4 ./ Zeitschrift fuer Krystallographie (149, 1979 - ) ZEKRD 209. 1994. P. 755. (Цит. по реферату)
26. Pujana A., Pizarro J.L., Goni A., Rojo T., Arriortua M.J.// Syntthesis and structural study of the Li1-3x Fex CoPO4 (x = 0 – 0,10) solid solution related to the litiophylite-triphylite family./ Anales de Quimica International Edition. 1998. P. 383-387. (Цит. по реферату)