| Скачать .docx |
Реферат: Эксклюзионная хроматография
Эксклюзионная хроматография
Основные понятия
Эксклюзионная хроматография представляет собой вариант жидкостной хроматографии, в котором разделение происходит за счет распределения молекул между растворителем, находящимся внутри пор сорбента, и растворителем, протекающим между его частицами.
В отличие от остальных вариантов ВЭЖХ, где разделение идет за счет различного взаимодействия компонентов с поверхностью сорбента, роль твердого наполнителя в эксклюзионной хроматографии заключается только в формировании пор определенного размера, а неподвижной фазой является растворитель, заполняющий эти поры. Поэтому применение термина «сорбент» к данным наполнителям в определенной степени условно.
Принципиальной особенностью метода является возможность разделения молекул по их размеру в растворе в диапазоне практически любых молекулярных масс – от 102 до 108 , что делает его незаменимым для исследования синтетических и биополимеров.
По традиции процесс, проводимый в органических растворителях, все еще часто называют гель-проникающей, а в водных системах – гель-фильтрационной хроматографией. Принят единый термин, который происходит от английского «Size Exclusion» – исключение по размеру – и в наиболее полной степени отражает механизм процесса.
Детальный разбор существующих представлений о весьма сложной теории процесса эксклюзионной хроматографии проведен в монографиях. Мы рассмотрим только принципиальные основы метода, достаточные для практической работы.
Объем эксклюзионной колонки можно выразить суммой трех слагаемых:
Vс=Vо+Vi+Vd,
где Vо – мертвый объем – объем растворителя между частицами сорбента (объем подвижной фазы); Vi– объем пор, занятый растворителем (объем неподвижной фазы); Vd– объем матрицы сорбента без учета пор.
Полный объем растворителя в колонке Vt (его часто называют полным объемом колонки, так как Vd не принимает участия в хроматографическом процессе) представляет собой суммy объемов подвижной и неподвижной фаз:
Vt=Vо+Vi.
Удерживание молекул в эксклюзионной колонке определяется вероятностью их диффузии в поры и зависит от соотношения размеров молекул и пор, что схематически показано на рис. 2.15. Коэффициент распределения Kd, как и в других вариантах хроматографии, представляет собой отношение концентраций вещества в неподвижной и подвижной фазах:
Кd = Сi / Со.
Так как подвижная и неподвижная фазы имеют одинаковый состав, то Kd вещества, для которого обе фазы одинаково доступны, равен единице. Эта ситуация реализуется для молекул с самыми малыми размерами (в том числе и молекул растворителя), которые проникают во все поры (см. рис. 2.15) и поэтому движутся через колонку наиболее медленно. Их удерживаемый объем равен полному объему растворителя Vt.

Рис. 1. Модель разделения молекул по размеру в эксклюзионной храматографии
Все молекулы, размер которых больше размера пор сорбента, не могут попасть в них (полная эксклюзия) и проходят по каналам между частицами. Они элюируются из колонки с одним и тем же удерживаемым объемом, равным объему подвижной фазы Vo. Коэффициент распределения для этих молекул равен нулю.
Молекулы промежуточного размера, способные проникать только в какую-то часть пор, удерживаются в колонке в соответствии с их размером. Коэффициент распределения этих молекул изменяется в пределах от нуля до единицы и характеризует долю объема пор, доступных для молекул данного размера. Их удерживаемый объем определяется суммой Vo и доступной части объема пор:
VR =Vo+KdVi.
Отсюда следует еще одно существенное отличие эксклюзионной хроматографии: в данном методе разделение заканчивается до выхода пика растворителя, в то время как в других вариантах ВЭЖХ компоненты смеси элюируются после пика растворителя.
Параметр k' в эксклюзионной хроматографии обычно не используют. Его можно выразить уравнением
k' = KdVi / Vo.
Для большинства современных сорбентов Vi ≈ Vo, поэтому k' ≈ Kd.
Связь между удерживаемым объемом и молекулярной массой (или размером молекул) образца описывается калибровочной кривой (рис. 2.16). Каждый сорбент характеризуется своей калибровочной кривой, по которой легко оценить область разделяемых на нем молекулярных масс. Точка А соответствует пределу эксклюзии, или мертвому объему колонки Vo. Все молекулы, масса которых больше, чем в точке А, будут элюироваться одним пиком с удерживаемым объемом Vo. Точка В отражает предел проникания, и все молекулы, масса которых меньше, чем в точке В, также будут выходить из колонки одним пиком с удерживаемым объемом Vt. Между точками А и В располагается диапазон селективного разделения. Соответствующий ему объем Vi=Vt-Vo часто называют рабочим объемом колонки. Отрезок CD представляет собой линейный участок калибровочной кривой, построенной в координатах VR–IgM. Этот участок описывается уравнением
Vr = C1 – C2lgM,
где C1 – отрезок, отсекаемый на оси ординат продолжением отрезка СD, C2 – тангенс угла наклона этого отрезка к оси ординат.
Величину С2 называют разделительной емкостью колонки; ее выражают числом миллилитров растворителя, приходящегося на один порядок изменения молекулярной массы. Чем больше разделительная емкость, тем селективнее разделение в данном диапазоне масс. В нелинейных областях калибровочной кривой (участки АС и BD) в связи с уменьшением С2 эффективность фракционирования заметно снижается. Кроме того, нелинейная связь между IgM и Vr существенно усложняет обработку данных и снижает точность результатов. Поэтому всегда нужно стремиться выбирать колонку (или набор колонок) так, чтобы разделение анализируемого полимера протекало в пределах линейного участка калибровочной кривой.
Если какое-либо вещество элюируется с удерживаемым объемом больше Vt, то это указывает на проявление других механизмов разделения (чаще всего адсорбционного). Адсорбционные эффекты обычно проявляются на жестких сорбентах, но иногда наблюдаются и на полужестких гелях, видимо, из-за повышенного сродства к матрице геля. Примером может служить адсорбция ароматических соединений на стирол-дивинилбензольных гелях.
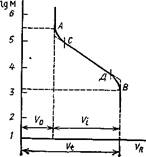
Рис. 2. Калибровочная кривая
Советские исследователи предложили теорию единого механизма жидкостной хроматографии полимеров на жестких гелях, из которой следует, что изменением параметров взаимодействия в системе полимер – сорбент – растворитель можно переходить от адсорбционного механизма к эксклюзионному и наоборот. В общем случае в эксклюзионной хроматографии нужно стремиться полностью подавить адсорбционные и другие побочные эффекты, так как они, особенно при исследовании молекулярно-массового распределения (ММР) полимеров, могут существенно исказить результаты анализа.
Принципиальными отличиями эксклюзионной хроматографии от других вариантов являются заранее известная продолжительность анализа в конкретной используемой системе, возможность предсказания порядка элюирования компонентов по размеру их молекул, примерно одинаковая ширина пиков во всем диапазоне селективного разделения и уверенность в выходе всех компонентов пробы за достаточно короткий промежуток времени, соответствующий объему Vt. Хотя данный метод применяют, главным образом, для исследования ММР полимеров и анализа макромолекул биологического происхождения (белки, нуклеиновые кислоты и т.д.), указанные особенности делают его чрезвычайно перспективным для анализа низкомолекулярных примесей в полимерах и предварительного разделения проб неизвестного состава. Получаемая при этом информация существенно облегчает выбор наилучшего варианта ВЭЖХ для анализа данной пробы. Кроме того, микропрепаративное эксклюзионное разделение часто используют в качестве первого этапа при разделении сложных смесей путем комбинации различных видов ВЭЖХ.
Ограниченный диапазон коэффициентов распределения определяет и главный недостаток эксклюзионной хроматографии – заметно меньшее, чем в других вариантах ВЭЖХ, число пиков, которые могут быть полностью разделены на колонке заданной эффективности. Однако в последнее время благодаря успехам достигнутым в технологии изготовления высокоэффективных колонок, этот метод все шире применяют и для разделения малых молекул.
Особенности аппаратуры
Аппаратура для эксклюзионной хроматографии принципиально ничем не отличается от той, которую используют в других видах ВЭЖХ. Эксклюзионное разделение можно осуществить на любом жидкостном хроматографе, установив в него соответствующие колонки. Характеристики аппаратуры влияют главным образом на точность получаемых результатов. Специфичными для данного метода являются только некоторые детекторы и особые требования к системам обработки данных.
Из всех вариантов ВЭЖХ в эксклюзионной хроматографии полимеров предъявляются наиболее жесткие требования к стабильности потока подвижной фазы. Поэтому нужно использовать насосные системы с точностью подачи не хуже 0,3–0,5%. В лучших насосах, разработанных специально для данного метода, нестабильность скорости потока снижена до 0,1%.
Между дозатором и колонками весьма желательно устанавливать фильтр с минимальным мертвым объемом, так как забивание входного фильтра колонки при анализе полимеров происходит гораздо чаще, чем в других видах ВЭЖХ.
Точность результатов в экоклюзионной хроматографии полимеров заметно зависит от температуры. При ее изменении на 10 °С ошибка определения средних молекулярных масс превышает ±10% [23]. Поэтому в данном варианте ВЭЖХ термостатирование разделительной системы обязательно.
Дозатор и колонки обычно размещают в одном термостате. Как правило, достаточна точность поддержания температуры ±1 °С в пределах до 80–100 °С. В некоторых случаях, например, при анализе полиэтилена и полипропилена, рабочая температура составляет 135–150 °С. Необходимо также принимать меры для предотвращения заметных изменений температуры в линии, соединяющей колонку с детектором. При рабочих температурах до 40–50 °С и длине линии 5–8 см ее целесообразно изготавливать из фторопластового капилляра с наружным диаметром около 1,5 мм, внутренним – 0,3 мм. При более высоких температурах требуется термостатирование капилляра.
Наиболее распространенным детектором в эксклюзионной хроматографии полимеров является дифференциальный рефрактометр. При работе с этим детектором следует помнить, что в диапазоне примерно до 5⋅103 –5⋅104 его сигнал зависит от молекулярной массы полимера. Поэтому при исследовании полимеров, содержащих значительное количество низкомолекулярных фракций, в процессе обработки результатов нужно вводить соответствующие поправки или, если это возможно, проводить специальную калибровку детектора. Из детекторов, разработанных специально для анализа полимеров, следует упомянуть вискозиметрический детектор и проточный лазерный нефелометр (детектор малоуглового лазерного светорассеяния). Эти детекторы в комбинации с рефрактометром или другим концентрационным детектором позволяют непрерывно определять молекулярную массу полимера в элюенте. При их использовании отпадает необходимость калибровки разделительной системы по исследуемому полимеру, но обработка информации может осуществляться только на ЭВМ. Вискозиметрический детектор, кроме того, является очень удобным прибором для исследования длинноцепной разветвленности синтетических полимеров.
Обычные электронные интеграторы, используемые в ВЭЖХ индивидуальных соединений, непригодны для обработки данных, получаемых при эксклюзионной хроматографии полимеров. Для этой цели используют мини-компьютеры, которые выполняют по специальным программам необходимые вычисления и выдают результаты опреде-
ления в виде средних молекулярных характеристик или кривых ММР. Современные приборы могут быть оснащены дополнительными устройствами для полной автоматизации анализа. Применение автоматических дозаторов в сочетании с мини-компьютером позволяет выполнять различные калибровки, выдавать в требуемой форме данные по ММР, проводить их статистический анализ без участия операторов.
Как отмечалось выше, в настоящее время анализ полимеров проводят в основном на обычной хроматографической аппаратуре. Однако существуют и специальные приборы, предназначенные преимущественно для определения ММР полимеров. К ним относится, в частности, микрогельхроматограф ХЖ-1309. Технические характеристики хроматографа приведены в приложении 14.6. Этот уникальный прибор оснащен высокочувствительным лазерным рефрактометром с вместимостью кюветы 0,1 мкл [24] и микроколонками диаметром 0,5 мм с эффективностью около 30 тыс. т. т./м. Продолжительность анализа составляет 5–10 мин, а расход растворителя – приблизительно 100 мкл на один анализ, что позволяет работать с особо дефицитными и сверхочищенными растворителями. Калибровку прибора и обработку результатов проводят на ЭВМ с пакетом программ, обеспечивающих выполнение любых расчетов, необходимых в эксклюзионной хроматографии полимеров.
Выбор сорбента
Выбор сорбентов, обеспечивающих оптимальные условия для решения конкретной аналитической задачи, проводят в несколько этапов. Первоначально на основе данных о химическом составе или растворимости анализируемых веществ устанавливают, какой вариант процесса следует применить – хроматографию в водных системах или в органических растворителях, что в значительной степени определяет тип необходимого сорбента. Разделение веществ низкой и средней полярности в органических растворителях можно успешно осуществить как на полужестких, так и на жестких гелях. Исследование ММР гидрофобных полимеров, содержащих полярные группы, чаще проводят на колонках со стирол-дивинилбензольными гелями, так как в этом случае практически не проявляются адсорбционные эффекты и не требуется добавка модификаторов к подвижной фазе, что значительно упрощает подготовку и регенерацию растворителя.
Для работы в водных системах используют главным образом жесткие сорбенты; иногда очень хорошие результаты удается получить на полужестких гелях специальных типов. Затем по калибровочным кривым или данным о диапазоне фракционирования, приведенным в гл. 4, выбирают сорбент нужной пористости с учетом имеющихся сведений о молекулярной массе образца. Если анализируемая смесь содержит вещества, отличающиеся по молекулярной массе не более чем на 2–2,5 порядка, то обычно удается разделить их на колонках с одним размером пор. При более широком диапазоне масс следует использовать наборы из нескольких колонок с сорбентами различной пористости. Ориентировочно калибровочную зависимость в этом случае получают сложением кривых для отдельных сорбентов.
На выбранных таким образом колонках выполняют пробный анализ и при необходимости вносят изменения в систему колонок для оптимизации разделения. Оптимизацию приходится проводить, если колонки не обеспечивают требуемого диапазона разделения или эффективности разделения.
Наиболее сложным является выбор системы колонок для разделения синтетических полимеров, имеющих широкое ММР. Обычно для этой цели применяли наборы из трех – пяти колонок, содержащих сорбенты с последовательно возрастающим размером пор
(например, µ-стирогель 102+103+104+105⊕)*, области разделения которых перекрываются. При этом, как правило, получали калибровочную зависимость с линейным диапазоном около трех порядков и с достаточно большими криволинейными участками, а продолжительность анализа (при наиболее типичной скорости потока 1 мл/мин) составляла 40–45 мин. Главным недостатком данной калибровки является существенное ухудшение селективности разделения на криволинейных участках, что заметно снижает точность результатов. Кроме того, резко усложняется обработка экспериментальных дан-ных. Поэтому особое значение имеет выбор такой разделительной системы, которая характеризуется линейной зависимостью логарифма молекулярной массы полимера от удерживаемого объема. В этом случае можно рассматривать хроматограмму как зеркальное отображение дифференциальной кривой ММР в логарифмическом масштабе.
Предложен принцип бимодального распределения размеров пор, который позволяет составлять наборы колонок с значительно лучшими рабочими характеристиками. В соответствии с этим принципом, для составления набора колонок с линейной калибровочной зависимостью в широком интервале молекулярных масс нужно использовать только два сорбента с размерами пор, отличающихся на один-полтора порядка и имеющих умеренно узкое распределение пор по размеру. Разделительная емкость С2 колонок с этими сорбентами должна быть примерно одинаковой. Полученные бимодальные наборы колонок, как правило, имеют линейный участок калибровочной кривой, перекрывающий около четырех порядков изменения молекулярной массы, и умеренную разрешающую способность. За счет сокращения числа колонок соответственно уменьшается продолжительность разделения. Так, бимодальные наборы, выпускаемые фирмой «Дюпон» и состоящие из колонок с зорбаксами PSM-60 и PSM-1000 длиной по 25 см, имеют линейную калибровку в диапазоне молекулярных масс от 2–102 до 106 и гарантированную эффективность не менее 20 000 т. т.
Дальнейшие исследования показали, что необходимое распределение пор сорбента по размерам, обеспечивающее линейную калибровку в любом заданном диапазоне молекулярных масс, в общем случае является мультимодальным и может быть рассчитано на ЭВМ.
Таким образом, имея несколько сорбентов разной пористости с известным распределением пор по размеру, можно рассчитать состав смешанного «линейного» сорбента.
Так, калибровочная зависимость тримодального сорбента на основе макропористых стекол в диапазоне молекулярных масс от 2–103 до 4–105 имела значительно более высокую степень линейности, чем у бимодальных наборов, описанных в работе.
Линейные наборы можно составлять и из колонок с полужесткими гелями. Приведем пример составления линейного набора из колонок с стирогелями в том случае, когда разделительная емкость первой колонки заметно меньше, чем второй. Полученная калибровочная зависимость представляет собой ломаную линию с точкой излома, соответствующей молекулярной массе около 20 000 (верхний предел селективного диапазона первой колонки). При подключении еще одной колонки с размером пор 500 Д разделительная емкость в диапазоне масс до 20 000 увеличилась примерно вдвое, а калибровочная зависимость набора стала практически линейной в диапазоне молекулярных масс от 5–102 до 2–106 .
По мнению Йоу с сотр., колонки с жесткими гелями лучше подходят для сочетания в бимодальные наборы, так как свойства этих сорбентов более стабильны от партии к партии. Однако следует отметить, что в последние годы качество колонок с полужесткими гелями и воспроизводимость их характеристик значительно улучшились. Отмечено, что современные полимерные сорбенты более однородны по размерам пор, чем сорбенты на основе силикагелей [28]. Опыт работы одного из авторов с колонками типа ц-сферогель показал, что соединение различных колонок требуемой пористости, взятых «через одну» (например, 5–102 +104 0 или 103 +105 0), в большинстве случаев позволяет получить линейную калибровочную зависимость в диапазоне около 4 порядков.
Оптимизацию разделительной системы целесообразно рассмотреть на примере исследования образца, о котором не известно ничего, кроме растворимости (в воде или в органическом растворителе). Предварительное разделение образца проводят на бимодальном наборе колонок с широким диапазоном разделения. В зависимости от вида получаемой хроматограммы (рис. 3) изменяют разделительную систему в соответствии с рекомендациями, приведенными ниже.
1. Набор оптимален.
2. Недостаточный диапазон разделения: имеются высокомолекулярные фракции, попадающие в область эксклюзии. Необходимо добавить колонку с более крупными порами.
3. Имеются фракции, попадающие в область эксклюзии; колонка II не участвует в разделении. Необходимо заменить ее на колонку с более крупными порами, чем у колонки I.
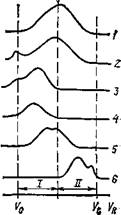
Рис. 3. Различные виды хроматограмм неизвестного образца: 1 – область разделения крупнопористой колонки; 2 – область разделения мелкопористой колонки
4. Колонка II не участвует в разделении. Следует использовать две колонки типа I.
5. Улучшение разделения можно получить на двух колонках со средним размером пор.
6. Недостаточная селективность разделения; колонка I не участвует в разделении.
Следует использовать две колонки типа II.
Если в любом из рассмотренных вариантов необходимо повысить эффективность разделения, то следует удвоить число колонок, входящих в выбранный набор.
Описанный процесс оптимизации не учитывает особенностей, связанных с адсорбцией анализируемых веществ и другими побочными явлениями. Устранение адсорбции за счет модификации подвижной фазы рассмотрено ниже.
Для предотвращения специфических затруднений, возникающих при эксклюзионной хроматографии биололимеров, витаминов и различных лабильных соединений, в последнее время чаще всего используют сорбенты с привитой карбогидратной (типа диол) или эфирной фазой (µ-бондагель Е) и полимерные сорбенты, предназначенные для работы в водных средах.
Выбор растворителя
Растворители, применяемые в эксклюзионной хроматографии, должны удовлетворять следующим основным требованиям:
1) полностью растворять образец при температуре разделения;
2) смачивать поверхность сорбента и не ухудшать эффективность колонки;
3) предотвращать адсорбцию (и другие взаимодействия) разделяемых веществ с поверхностью сорбента;
4) обеспечивать максимально высокую чувствительность детектирования;
5) иметь низкую вязкость и токсичность.
Кроме того, при анализе полимеров имеет существенное значение термодинамическое качество растворителя: весьма желательно, чтобы он был «хорошим» по отношению к разделяемому полимеру и матрице геля. Свойства, наиболее распространенных растворителей для эксклюзионной хроматографии приведены в приложении 2.
Растворимость образца обычно является главным лимитирующим фактором, ограничивающим ассортимент пригодных подвижных фаз. В приложении 3 указаны основные типы полимеров, которые целесообразно анализировать в тех или иных растворителях.
Наилучшим органическим растворителем для эксклюзионной хроматографии синтетических полимеров по комплексу свойств является тетрагидрофуран. Он обладает уникальной растворяющей способностью, низкой вязкостью и токсичностью, лучше многих других растворителей совместим со стирол-дивинил-бензольными гелями и, как правило, обеспечивает высокую чувствительность детектирования при использовании рефрактометра или УФ-детектора в области до 220 нм. Для анализа высокополярных и нерастворимых в тетрагидрофуране полимеров (полиамиды, полиакрилонитрил, полиэтилен-терефталат, полиуретаны и др.) обычно используют диметилформамид или м -крезол, а разделение полимеров низкой полярности, например различных каучуков и полисилоксанов, часто проводят в толуоле или хлороформе. Последний является также одним из лучших растворителей при работе с ИК-детектором. о -Дихлорбензол и 1,2,4 – трихлор-бензол применяют для высокотемпературной хроматографии полиолефинов (обычно при 135 °С), которые в других условиях не растворяются. Эти растворители имеют очень высокий показатель преломления, поэтому иногда их целесообразно использовать вместо тетрагидрофурана для анализа полимеров с низким коэффициентом преломления, что позволяет повысить чувствительность при детектировании рефрактометром.
Для предотвращения окисления растворителей и полужестких гелей в условиях высокотемпературной эксклюзионной хроматографии к о -дихлорбензолу и 1,2,4 – трихлор-бензолу добавляют антиокислители – 1,3 г/л 2,6 – ди-mреm -бутил-4-метилфенола (алко-фен БП, ионол) или 0,4 г/л 4,4/ -тиo-биc (6-трет -бyтил-3-метилфенол) а (тиоалкофен БМ, сантонокс R).
Жесткие сорбенты совместимы с любыми подвижными фазами, имеющими рН<8–8,5; при более высоких значениях рН силикагель начинает растворяться и колонка необратимо теряет эффективность. Стирол-дивинилбензольные гели совместимы в основном с
элюентами умеренной полярности. Для работы на колонках с µ-стирогелем (от 103 ⊕ и выше), по данным фирмы «Уотерс», пригодны тетрагидрофуран, ароматические и хлорированные углеводороды, гексан, циклогексан, диоксан, трифторэтанол, гексафтор-пропанол и диметилформамид.
Степень набухания частиц геля в различных растворителях неодинакова, поэтому замена элюента в колонках с данными сорбентами может привести к снижению эффективности за счет изменения объема геля и образования пустот. При использовании неподходящих растворителей (ацетон, спирты) происходит столь сильная усадка геля, что колонка оказывается безнадежно испорченной. У сорбентов с малым размером пор (типа µ-стирогеля 100⊕ и 500⊕) такая усадка наблюдается как в полярных, так и в неполярных растворителях, поэтому с ними, кроме того, нельзя работать в насыщенных углеводородах, фторированных спиртах и диметилформамиде. Удобным, хотя и весьма дорогим выходом из положения является использование отдельных наборов колонок для каждого применяемого растворителя. Некоторые фирмы с этой целью выпускают колонки с одним и тем же размером пор, заполненные разными растворителями – тетрагидро-фураном, толуолом, хлороформом и диметилформамидом.
Для предотвращения усадки геля рекомендуется следующий способ замены растворителя: колонку, содержащую растворитель А , присоединяют к насосу, устанавливают скорость потока растворителя А 0,3–0,5 мл/мин, проводят градиентное изменение состава элюента от 0 до 100% растворителя В со скоростью 1%/мин и промывают с той же скоростью растворителем В в течение 1,5–2 ч. Резкое изменение полярности растворителя почти всегда приводит к ухудшению характеристик колонки, и его лучше вообще избегать. Если такая замена все же необходима, то следует использовать промежуточный растворитель. Так, толуол сначала заменяют на тетрагидрофуран и через 1–2 дня – на диметилформамид. Падение эффективности при этом будет меньше, чем при одностадийной замене растворителя. В любом случае замена подвижной фазы в высоко-
эффективных колонках с полужесткими гелями остается весьма деликатной операцией, требующей особой осторожности. В принципе возможно приготовление таких колонок и для работы с полярными растворителями. Нужный растворитель при этом используют при набивке колонки. Однако вследствие малой набухаемости гелей в этих растворителях получаемые колонки обычно имеют более низкую эффективность.
При разделении макромолекул основной вклад в размывание полосы определяется затрудненной массопередачей. К сожалению, многие из применяемых элюентов имеют высокую вязкость. Для снижения вязкости (а также для улучшения растворимости) экс-клюзионную хроматографию часто проводят при повышенных температурах, что существенно улучшает эффективность хроматографической системы.
Анализ большинства полимеров на жестких гелях часто осложняется их адсорбцией. Для подавления адсорбции обычно используют растворители, которые адсорбируются на насадке колонки сильнее, чем анализируемые вещества. Если по каким-либо причинам это невозможно, то подвижную фазу модифицируют добавкой 0,1–2% полярного модификатора, например тетрагидрофурана. Значительно более сильными модификаторами являются этиленгликоль и полигликоли с различной молекулярной массой (ПЭГ-200, ПЭГ-400, карбовакс 20 М). Иногда, например при анализе поликислот в диметилформамиде, требуется добавка достаточно сильных кислот. Следует отметить, что полностью устранить адсорбцию добавкой модификаторов удается не всегда. В таких случаях нужно использовать полужесткие гели. Некоторые полимеры хорошо растворяются только в высоко полярных растворителях (ацетон, диметилсульфоксид и т.п.), несовместимых со стирол-дивинилбензольными гелями. При их разделении на жестких сорбентах выбор растворителя проводят в соответствии с общими принципами, изложенными выше.
Иная ситуация имеет место при проведении эксклюзионной хроматографии в водных средах. Из-за специфических особенностей многих разделяемых систем (белки, ферменты, полиэлектролиты и др.) и разнообразия применяемых сорбентов существует очень много вариаций состава подвижной фазы для подавления различных нежелательных эффектов. Общими приемами модификации является добавка различных солей и применение буферных растворов с определенным значением рН. В частности, поддержание рН=<4 дает возможность подавить слабую ионообменную активность силикагелей, обусловленную присутствием на их поверхности кислых силанольных групп. Требуемая ионная сила подвижной фазы достигается при концентрации буферного раствора 0,05–0,6 М; оптимальную концентрацию подбирают экспериментально. Для предотвращения ионообменной сорбции катионных соединений наиболее часто используют такой активный модификатор, как тетраметиламмонийфосфат при рН≅3. Однако при разделении некоторых белков могут проявляться гидрофобные взаимодействия, в свою очередь осложняющие эксклюзионный механизм разделения. Те же эффекты иногда проявляются и при работе с дезактивированными гидрофильными сорбентами. Для их устранения к растворителю добавляют метанол. Иногда в водную подвижную фазу вводят полярные органические растворители, полигликоли, кислоты, основания и поверхностно-активные вещества.
Литература
1. Aleksandrov M.L. e. a./J. High Resol. Chromatogr. Commun, 1983, v. 6, No. 11, p. 629–631.
2. Yau W., Grinnard G., Kirkland /./J. Chromatogr., 1978, v. 149, p. 465.
3. Виленчик Л. 3. и dp. / Высокомол. соед., 1980, т. А22, №11, с. 2801–2804.
4. Нестеров В.В. и dp. / Высокомол. соед., 1984, т. Б26, №3, с. 163–167.
5. Benson J.R., Woo D. /./J. Chromatogr. Sci, 1984, v. 22, No. 9, p. 386 – 399.
6. Zdanov S.P. e. a./J. Chromatogr, 1973, v. 77, No. 2, p. 149–159.
7. Schlechter /./Anal. Biochem, 1974, v. 58, No. 1, p. 30.
8. Нефедов П.П. и dp. / Высокомол. соед, 1981, т. А23, №5, с. 943–950.
9. Nakamura К., Endo R./J. Appl. Polym. Sci, 1981, v. 26, p. 2657–2664.
10. Ициксон Л.Б., Филиппов А.А. // В кн.: Ill Всесоюзный симпозиум по молекулярной жидкостной хроматографии, тезисы докладов. Рига, изд. Института органич. синтеза АН Латв. ССР, 1984, с. 126–127.