| Скачать .docx |
Реферат: Активация малых молекул
1. Введение
В катализе часто применяют термин “активация”, понимая при этом повышение реакционной способности реагентов. Попытаемся наполнить этот термин конкретным физическим содержанием. Под активацией субстрата будем понимать степень и характер воздействия катализатора на субстрат, приводящих к превращению первичного комплекса в промежуточное соединение, содержащее в координационной сфере реакционноспособные фрагменты субстрата. Анализ имеющихся данных о влиянии координации на физические свойства лигандов и о реакциях координированных лигандов позволяет понять наиболее существенные черты механизма активации каждой молекулы.
Рассмотрим современные представления о механизме активации простых молекул комплексами переходных металлов. Механизмы активации молекул различного типа кислотными катализаторами рассмотрены ранее.
Сущность активации заключается в изменении определенных характеристик субстратов при образовании первичного комплекса с катализатором.
Кат-р + субстрат ® Х1 ® Х2 ….. ® Хn ® P + K
При образовании первичного комплекса возможны три варианта
Образуется очень прочный комплекс между субстратом и катализатором. Субстрат в таком комплексе оказывается менее реакционноспособным, чем в свободном виде.
В первичном комплексе происходит изменение характеристик субстрата в желательном направлении: изменение валентных углов, понижение частоты валентных колебаний и удлинение связей. Реакционная способность увеличивается.
При комплексообразовании происходит расщепление субстрата на фрагменты, часть которых или все могут оказаться в координационной сфере комплекса.
Второй и третий случаи называют активацией за счет координации и активацией за счет присоединения, соответственно.
2. Активация молекулы водорода
Проблема активации этой молекулы важна т.к. водород один из основных реагентов НХС и ООС (гидрообработка, гидрогенолиз, гидрирование ненасыщенных соединений, синтезы из синтез-газа).
Молекула Н2 – слабый донор (IH2= 15,4 эВ) и слабый акцептор (FH2= -0,7 эВ).
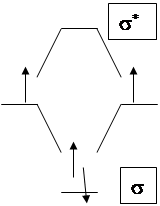
Рассмотрим возможности активации этой молекулы с точки зрения теории МО.
 |
|
|
|
Энергия диссоциации связи Н-Н зависит от заселенности орбиталей электронами (табл. 1).
Таблица 1
Энергия диссоциации связи Н-Н в зависимости от заселенности орбиталей
| Частица | ЕD, кДж/моль | LH-H, Å |
| Н2 | 430,5 | 0,74 |
| Н2+ | 259,2 | 1,07 |
| Н2 - | 17,14 | 0,86 |
|
Хотя сродство Н2 к Н+ довольно велико (в газовой фазе DНО = -322кДж/моль) активация протоном молекулы Н2 (вариант а) оказалась возможной только в растворе суперкислоты SbF5-HF в апротонных средах (SO2, SO2ClF ), cудя по реакциям дейтерообмена. Вариант (б) оказался более простым. Активация водорода легко осуществляется основаниями (ОН-, КNH2, К). Промотирование электронов на разрыхляющую орбиталь водорода является решающим фактором активации (К+Н2-).
Первичные комплексы для водорода были получены примерно в 1980 г. Первым из них был (CO)3W(P i-Pr3)2(h2-H2). Структура комплекса представляет собой октаэдр с молекулой водорода, занимающей одно координационное место в экваториальной плоскости вместе с тремя карбонилами. Длина связи Н-Н составляет 0,84 Å (сравни с табл 1). В аналогичном комплексе иридия Ir(H)2(h2-H2)[P(C6H11)3]2 наблюдается быстрый обмен между координированным молекулярным водородом и гидридными лигандами. Однако в этих комплексах координирована молекула водорода и ее активация сводится лишь к удлинению связи H-H.
В большинстве случаев взаимодействие водорода с комплексами переходных металлов приводит к разрыву связи в молекуле водорода. При этом возможны два основных направления превращения водорода при взаимодействии с комплексами переходных металлов.
В случае комплексов металлов в высоких степенях окисления с преобладанием акцепторных свойств происходит гетеролитический разрыв связи в водороде (электрофильное замещение Н+ на комплекс металла).
Cu2+ + H2 ® CuH+ + H+
PtCl2 + H2 ® ClPtH + HCl
RuCl63- + H2 ® H RuCl53- + HCl
Комплексы металлов в низших степенях окисления, имеющие возможность повысить степень окисления и координационное число, ведут к гомолитическому расщеплению связи Н-Н. В этом случае дативная компонента связи преобладает над донорно-акцепторной.
IrCl(CO)L2 + H2 ® (H)2 IrCl(CO)L2
Co2(CO)8 + H2 ® 2 HCo(CO)4
2 Co(CN)53-+ H2 ® 2 HCo(CN)53-
В любом из перечисленных вариантов образуются гидридные комплексы переходных металлов. Комплексы такого типа предполагаются в качестве катализаторов и интермедиатов многих процессов с участием водорода.
4. Активация молекулы монооксида углерода
Гетероатомная молекула оксида углерода имеет очень большую энергию связи 256 кКал/моль. Она слабый донор и сильный p-акцептор (акцептирует на разрыхляющие орбитали). Относительное расположение молекулярных орбиталей для СО имеет особенность. Pz уровень в атоме кислорода расположен ниже, чем соответствующий. уровень для атома углерода (из-за большего заряда ядра), поэтому он (Pz уровень в атоме О) сильно взаимодействует с 2s–орбиталью атома углерода. В связи с этим Pz–молекулярная орбиталь СО располагается выше, чем вырожденные Pх и Pу орбитали (см. диаграмму). Пара электронов, которая располагается на Pz орбитали, является несвязывающей и локализована на атоме углерода, а пара электронов на 2s–орбитали - также несвязывающая и локализована на атоме кислорода. Причем, Pz – молекулярная орбиталь имеет в основном р-характер с большим лепестком, вытянутым от связи С-О. Вакантные орбитали (Pz*, Pх*, Pу*) также локализованы на атоме углерода, поэтому практически во всех случаях оксид углерода координируется через углерод.
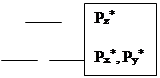 |
||||||
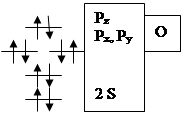 |
||||||
|
||||||
|
||||||
За счет заполненной Pz-орбитали, локализованной на углероде, СО обладает слабыми донорными свойствами и образует донорно-акцепторную компоненту связи, взаимодействуя с подходящей по симметрии вакантной орбиталью dz2 переходного металла. За счет вакантных разрыхляющих Px и Py орбиталей у СО есть возможность проявлять акцепторные свойства. Две разрыхляющие p-орбитали по симметрии могут взаимодействовать с заселенными dxy и dxz орбиталями переходного металла.
Из всего вышесказанного следует, что СО является s-лигандом, но в подавляющем большинстве случаев его следует рассматривать одновременно как s-донор и p-акцептор с преобладанием акцепторных свойств.
Карбонильные комплексы известны для большинства переходных металлов. Первые комплексы были получены в конце 19-го века. Например [Pd(CO)X]n, [Pt(CO)X]n, Co2(CO)8, Ni(CO)4. Координация оксида углерода в комплексах бывает концевая (терминальная) и мостиковая (с участием двух атомов металла или трех атомов металла).
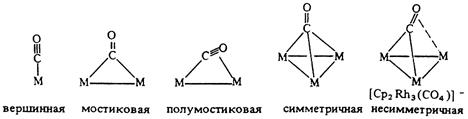
Первый тип координации является концевым и реализован во многих моно- и полиядерных комплексах, например в тетраэдрическом моноядерном тетракарбониле никеля или биядерном дикобальтоктакарбониле.
Следующие типы координации - симметричная и несимметричная мостиковая (µ- и µ3-тип) - широко распространены в химии кластеров.
Так, в кластере состава
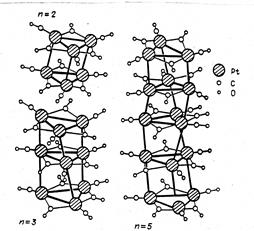
Pd4(CO)4(OAc)4 имеются только µ-СО-группы. В кластере [Pt3(CO)6]2-n наблюдаются два типа координации СО – и концевые, и мостиковые лиганды СО, причем в каждом металлотреугольнике Pt3 представлены по три лиганда обоих типов координации.
Примеры мостиковой координации СО-группы по µ- и µ3-типу реализуются в анионном гетероядерном комплексе состава Na2{Pd4[CpMo(CO)3]4}. Здесь на каждом металлотреугольнике Pd2Mo одна СО-группа координирована по µ3-типу, а две другие – по µ-типу по ребрам Pd-Mo, причем последние СО-группы слегка асимметричные (связь Mo-C короче связи Pd-C).
Pd4[CpMo(CO)3]42- |
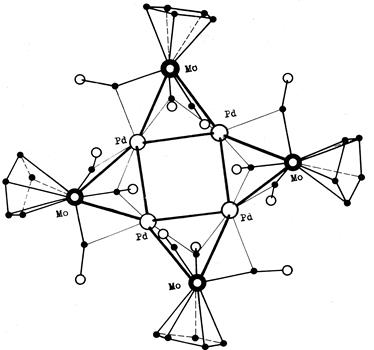
Следует добавить, что при терминальной координации карбонил является донором 2 электронов. Эта же донорная способность сохраняется в том случае, если СО координирован мостиком, но при этом направление связи С-О остается перпендикулярным ребру (или грани), на которой он координирован. В противном случае в связь с металлом начинают включаться электроны кратной связи и атома кислорода.
Как уже отмечалось выше, координация СО по обоим атомам возможна, хотя и встречается относительно редко. При такой координации происходит значительное удлинение связи С-О (от 1.13 до 1.30 А). Примеры такой координации приведены ниже.
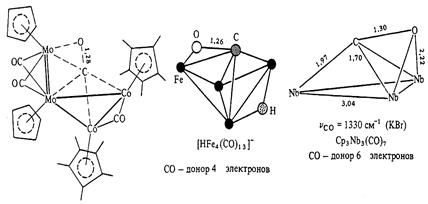
Если в образовании связи металл-лиганд участвуют оба атома, то СО-группа является донором 4 электронов. В случае, когда молекула СО расположена параллельно плоскости М3-цикла, она становится донором 6 электронов.
И, наконец, линейная координация СО отмечена в металлоорганических соединениях металлов начала больших периодов периодической системы:
(CO)5V-C-O-V-O-C-V(CO)5 или (η5-С5Me5)(Me)Ti-O-C-Mo(CO)3Cp
![]()
![]() Координационная связь между СО и переходным металлом складывается из двух компонент – донорноакцепторной ( СО – донор, металл – акцептор за счет вакантных орбиталей, например, dz2 и dx2-y2 для октаэдрических комплексов) и дативной ( металл – донор за счет заполненных dxy, dxz, dyz, СО – акцептор за счет вакантных орбиталей. Обе компоненты способствуют ослаблению связи С – О. В зависимости от степени заселения разрыхляющих орбиталей СО происходит большее или меньшее увеличение длины, понижение частоты валентных колебаний и уменьшение энергии связи С – О ( табл.)
Координационная связь между СО и переходным металлом складывается из двух компонент – донорноакцепторной ( СО – донор, металл – акцептор за счет вакантных орбиталей, например, dz2 и dx2-y2 для октаэдрических комплексов) и дативной ( металл – донор за счет заполненных dxy, dxz, dyz, СО – акцептор за счет вакантных орбиталей. Обе компоненты способствуют ослаблению связи С – О. В зависимости от степени заселения разрыхляющих орбиталей СО происходит большее или меньшее увеличение длины, понижение частоты валентных колебаний и уменьшение энергии связи С – О ( табл.)
| Молекула или ион | Энергия связи С-О, кКал/моль | Частота валентных колебаний, см-1 | Длина связи С-О, Å |
| СО (некоорд. молекула) | 256 | 2143 | 1,128 |
| СО+ | 2214 | 1,115 | |
| СО (коорд. концев.) | 2214 ¸1980 | ||
| СО (мостиков. ) | 1970 ¸ 1650 | ||
| Ni(CO)4 | 2057 | 1,15 | |
| H2CO | 170 | 1750 | 1,21 |
Из приведенных данных видно, что при координации происходит изменение свойств молекулы СО (см. выше) и координированная молекула по характеристикам приближается к карбонильной группе органических соединений. А в этом и заключается цель большинства хических процессов органического синтеза с участием СО. Изменение характеристик координированного СО приводит к изменению реакционной способности.
Наиболее характерная реакция координированного СО – реакция с нуклеофилами с образованием ацильного металлоорганического соединения:
Образование интермедиатов такого типа предполагается в процессах карбонилирования алкинов, окислительного карбонилирования спиртов, алкенов и алкинов.
Изложенная схема активации применима к большинству субстратов: алкенам, диенам, алкинам, ароматическим соединениям, СО2.
Превращения олефинов и ацетиленов относятся к важнейшим реакциям основного органического синтеза. Как правило, эти превращения включают активацию π-лигандов в π-комплексах переходных металлов. Фактически, в рамках тех понятий и терминов, которые мы обсуждаем, π-комплексы являются первичными комплексами.
π-Комплексы моноолефинов, сопряженных и несопряженных диолефинов и ацетиленов известны почти для всех переходных металлов. При этом встречаются моно-, би- и полиядерные соединения. Известны также соединения, содержащие 2, 3 и даже 4 молекулы олефина на атом металла, например, AcacRh(C2 H4 )2 , Ni(C2 H4 )3 и Ir(C2 H4 )4 Cl.
В ацетиленовых π-комплексах состава Mm (C2 R2 )n соотношение m/n меняется весьма широко. Например, известны комплексы состава Co4 (CO)10 (C2 H2 ), Pt(C2 R2 )2 , W(CO) (C2 R2 )3 .
Координация всех π-лигандов сопровождается более или менее значительными изменениями их физических характеристик – понижается частота валентных колебаний кратных связей, увеличивается длина связей С-С, изменяются величины валентных углов.
Природа химической связи в π-комплексах переходных металлов имеет много общего. Рассмотрим основные положения на примере π-комплексов олефинов и ацетиленов.
Молекула этилена по величине потенциала ионизации не отличается от молекулы аммиака (IC 2 H 4 = 10.5 эв). Донорные свойства ацетилена выражены несколько слабее (IC 2 H 2 = 11.4 эв). В ацетилене, однако, нижние вакантные МО лежат ниже, чем у этилена, поэтому молекула ацетилена характеризуется более выраженными акцепторными свойствами.