| Скачать .docx |
Реферат: Decomposition du percarbonate de 0,O-t-butyle et 0-isopropbnyle en solution: acetonylation des esters, acides et nitriles
Decomposition du percarbonate de 0, O-t-butyle et 0-isopropbnyle en solution: acetonylation des esters, acides et nitriles
The Free-Radical Decomposition of 0,O-t- Butyl and 0-Isopropenyl Peroxycarbonate in Solution: the Acetonylation of Esters, Acides and Nitriles
Summary
The free-radical decomposition of 0.0- t- butyl and 0 -isopropenyl peroxycarbonate in substrates possessing mobile H-atoms (S—H) consists mainly in an induced chain process leading to acetonylated derivatives of the solvent. Fairly good yields are obtained but the acetonylation of functional substrates often gives mixtures of isomers.
In the case of methyl acetate, the acetonylation occurs on the C-atoms adjacent to the carboayl (acyloxy moiety) and to the 0-atom (alkoxy moiety). However, the relative amounts of the isomeric products depend on the concentration of the peroxycarbonate solutions; at lowest concentration, methyl 4-oxopentanoate (acyloxy moiety) is obtained selectively. It is assumed that the free radicals issued from the solvent are able to abstract H-atoms of other molecules of solvent before adding to the double bond of the peroxycarbonate; the more the peroxycarbonate solution is diluted the more the transfers from the C-atom adjacent to the carbonyl to the radicals adjacent to the 0-atom are favoured. In the case of methyl alkanoates, H-transfers from the a-C-atoms to j? -radicals of the acyloxy moiety may account for the orientation of the process. Owing to similar H-transfer processes, the acetonylation of functional esters, of acids and nitriles is selective in most cases.
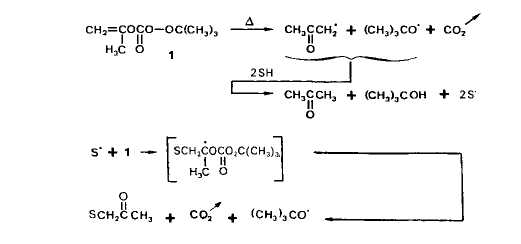
La decomposition du percarbonate de 0. 0-t-butyle et 0-isopropenyle (1) en solution dans de nombreux composes (S-H) possedant des atomes d'hydrogene labiles permet de rtaliser Г acetonylation radicalaire de ces substrats [11. Cette reaction resulte de I'addition de radicaux libres issus du solvant (S‘) sur la double liaison du percarbonate et de la dtcomposition qu'elle induit; les radicaux S' sont eux-m2mes form& au cours de processus d'arrachement d'hydroghe, notamment par les radicaux t-butoxyle (Schkma I).
A la suite de l'ttude generale, nous avons precise le comportement des radicaux correspondant aux cyclanes [2], aux cyclanones et oxacyclanes [3], ... en prtsence du percarbonate 1, et envisage dans quelles conditions la substitution pouvait 2tre orientke stlectivement vers certains sites rtactifs des moltcules.
Nous ktudions ici I'acttonylation des acides et de leurs derives. Les esters sont u priori susceptibles de soulever des problemes particuliers. En effet, les deux types d'ato-mes d'hydrogene actives, en a du carbonyle et en n de l'oxygene, rendent possible la competition entre sites reactifs et donc des variations dans l'orientation de l'acttonyla-tion en fonction des conditions exptrimentales. Le probleme peut se compliquer a la suite d'tventuelles competitions entre positions a, p voire y par rapport au groupe ou B l'atome activant; pour les acides et les nitriles, seule une concurrence entre sites reactifs de ce type doit, bien siir, &tre envisagee.
AcCtonylation des esters. - Cas de Г acetate de mithyle. Desirant comparer les rCac-tivitis apparentes des fragments acyloxyle et alcoxyle des molecules d'esters (d'aprks Ies quantitks des dkrivts acCtony1i.s formkes par reaction avec le percarbonate l), nous avons choisi comme modele l'acetate de mtthyle (2) qui ne possede que des sites rkactifs en a du groupement fonctionnel; son principal inconvenient est une riactivite plu-t6t faible vis 5 vis des radicaux t-butoxyle [4].
Rksultuts. Les conditions exptrimentales (chauffage pendant 2,5 h a 130“C) ont kt6 choisies pour avoir une rkaction totale du percarbonate mis en jeu et nous avons optrt avec des quantites relatives variables de percarbonate 1 et d'acktate 2. Dans tous les cas, nous avons observe la formation de deux derives acetonylks, 2'a (c8te acyloxyle) et 2'b (cGte alcoxyle), et de produits secondaires: t-butanol, acetone, acetonylacktone et dehydrodimlres de l'acttate de methyle.
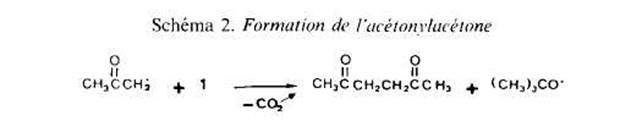
Le Schema I rend compte de la formation du t-butanol et d'une partie de l'acktone, l'autre partie de cette derniere provenant, avec les radicaux mtthyle, de reactions de scission en p de radicaux t-butoxyle (eu tgard a la faible reactivitk de I'acetate de methyle, cette scission se produit de maniere importante; le rendement en t-butanol est faible - 50% environ de la quantiti maximale theorique). L'acktonylacttone provient de l'addition de radicaux acktonyle, issus de la decomposition du percarbonate, sur la double liaison de ce dernier (Schima 2) ; aucun produit correspondant a l'addition d'autres radicaux, t-butoxyle ou mtthyle en particulier, n'a Ctk identifie dans les melanges reactionnels. Les dehydrodim2res de Гacetate de mkthyle, enfin, sont des produits de terminaison, par couplage de radicaux S’.
Les rendements en 0x0-Cpentanoate de methyle (2'a), et acttoxy-4-butanone-2 (2'b) sont tr&s modestes dans toutes les conditions (25% au mieux, par rapport a la quantite de 1 mise en jeu, dans le cas d'un rapport molaire initial 2/1 kgal a 50:1, les rendements s'entendant pour l'ensemble des isomires 2'a et 2'b isoles des melanges reactionnels par distillation). Les resultats, consignes dans le Tableau I, montrent que la sklectivite de l'acetonylation en a du carbonyle (isomire 2'a) augmente quand on op&re avec des solutions de plus en plus dilukes en percarbonate 1 dans le substrat.
Tableau 1. Orientation de I'acCtonylation de I hcQtate de mQthyle (2) par action du percarbonate 1 en fonction de la concentration de 1 dans 2
![]() 112
1:lO 1:20 1:50 1:100 1:150
112
1:lO 1:20 1:50 1:100 1:150
![]() 2'a/2'b
55:45 66:34 15:25 87:13 98:2
2'a/2'b
55:45 66:34 15:25 87:13 98:2
Discussion. Pour expliquer ce resultat il n'est pas possible d'invoquer une evolution avec la concentration initiale des solutions, ni de la rCactivitC des radicaux t-butoxyle, ni de l'aptitude a l'addition des radicaux 2.a ou 2.b. Notre hypothirse est alors qu'il se produit, en competition avec l'addition des radicaux S' sur la double liaison du percarbonate, des transferts homolytiques d'hydrog6ne (nous utiliserons par la suite le vocable abrege de transferts) entre les atomes de carbone en a du groupement fonctionnel de molecules de substrat et les radicaux 2.a et 2.b.
Les differentes possibilites d’evolution des radicaux sont representees dans le schema 3.
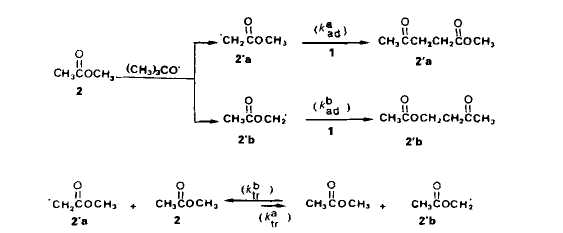
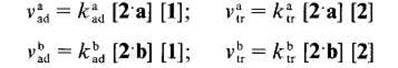
I1 est possible dexprimer les vitesses des &tapes Cltmentaires d'addition et de trans-fert. Nous disposons de peu d'kltments nous permettant de connaitre la repartition initiale entre radicaux 2.a et 2.b. En tenant compte des energies de liaison (effets enthalpi-ques), les radicaux 2 a devraient Ctre preponderants [5] mais, en se basant sur le caractere electroaccepteur des radicaux t-butoxyle [6] et sur le fait que les positions en a du carbonyle sont beaucoup plus appauvries en electrons que celles en a de l'oxygene, ce sont les rddicaux 2.b qui pourraient Ctre majoritaires. En outre les radicaux nikthyle, issus de la scission en ft de radicaux t-butoxyle, ont un caractere nucltophile [7] et en depit de leur modeste rkactivitk [8], ils peuvent contribuer a crker des radicaux 2.a. Quoi qu'il en soit, la repartition initiale a peu d'importance puisqu'elle va Ctre modifiee par les processus de transfert et que la repartition des produits de reaction ne sera pas le reflet de I'attaque initiale sur les molecules de substrat.
En ce qui concerne l’kvolution des radicaux 2.a et 2.b, il n'est pas tellement surpre-nant que les orientations soient differentes. Pour les radicaux 2,a, les resultats experi-mentaux montrent que le transfert ne se manifeste pas mCme lorsque, par effet de dilution, la concentration en substrat 2 est augmentee dans des proportions importan-tes. Rendus fortement klectrophiles par l'effet electroaccepteur marque du carbonyle, les radicaux 2.a sont probablement plus aptes a s'additionner sur la double liaison qu'a attaquer l'atome de carbone situe en a de l'oxygene. Pour les radicaux 2.b, la constata-tion experimentale est inverse, la proportion du transfert par rapport a l'addition aug-mentant avec la concentration en substrat. Ici, l'absence de preference pour l'addition semble pouvoir Ctre rapprochee du caractere neutre ou, au plus, faiblement klectrophile du radical en a de I'oxygene (contrairement au cas des radicaux isomires en a du carbonyle, aucune dklocalisation de l'electron ctlibataire ne peut ttre envisagee). Ainsi, l'accroissement de la sklectiviti de l'acktonylation en a du carbonyle provoquke par diminution de la concentration initiale des solutions de percarbonate 1 dans le substrat est en relation avec le caractere polaire plus ou moins marque des radicdux issus de ce substrat.
Notre argumentation implique que l'addition des radicaux 2.a, electrophiles, sur la double liaison du percarbonate soit plus facile que celle des radicaux 2.b, nettement moins electrophiles. On arrive ainsi a la conclusion que la double liaison du percarbonate possede plutbt un ex& d'electrons ce qui ne peut se comprendre, en tenant compte de l'effet donneur moyen du substituant methyle, qu'en admettant que la fonc-tion percarbonate n'attire les electrons que de faqon negligeable.
Cas des alcanoates de mkthyle. Ayant mis en kvidence, avec l'acetate de mkthyle, l'existence de transferts d'hydrogine de la partie acyloxyle vers la partie alcoxyle, il nous a paru interessant de comparer les rkactivitts apparentes des atomes de carbone en a, p, ... du carbonyle. La comparaison pour les atomes de carbone en a, p, ... de l'atome d'oxygene n'aurait pas de sens en raison des transferts. Nous avons etudie l'acetonylation, par action du percarbonate 1, des propionate (3), butyrate (4) et isobu-tyrate (5) (Schgma 4).
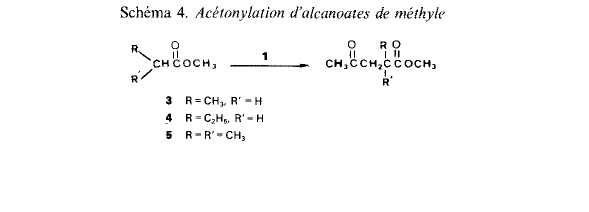
![]() Pour des reactions effectuees dans les conditions deja prkcisees (2,5 h a 130°C) et avec un rapport molaire initial percarbonate/substrat Cgal a 1:50 (rapport donnant les meilleurs rendements en derives acetonyles), nous avons obtenu 1es rtsultats decrits dans le Tableau 2.
Pour des reactions effectuees dans les conditions deja prkcisees (2,5 h a 130°C) et avec un rapport molaire initial percarbonate/substrat Cgal a 1:50 (rapport donnant les meilleurs rendements en derives acetonyles), nous avons obtenu 1es rtsultats decrits dans le Tableau 2.
Tableau 2. Acttonylation d'alcnnoates de mPrhylr
Substrat RdtYo % de derives acetonylks «cote»
![]() alcoxyle acyloxyle acyloxyle
alcoxyle acyloxyle acyloxyle
carbone a carbone B
3 ![]() 51 2 98 0
51 2 98 0
4 58 traces 80 20
5 15 0 100 0
On constate que les rendements en derives acktonylks (Oh par rapport a 1) augmen-tent considkrablement lorsque l'atome de carbone en a du groupe fonctionnel est se-condaire (esters 3 et 4) au lieu de primaire (ester 2). Ceci est en accord avec l'observa-tion genkrale que l'arrachement des atomes d'hydrogine par des radicaux libres est beaucoup plus facile sur les sommets secondaires que sur les primaires [9]. Cependant, pour l'isobutyrate de methyle (5), le rendement est faible en depit de la grande reacti-vitk attribuee aux sommets tertiaires [9]. Dans ce cas, on peut supposer que c'est I'addi-tion sur la double liaison du percarbonate du radical tertiaire en a d'un groupe ester qui est difficile, une confirmation a cette hypothese etant que l’on peut isoler dans les melanges rkactionnels des quantitts non negligeables du dkhydrodimere de I’ester cor-respondant au couplage de radicaux tertiaires.
Une seconde remarque est qu'il ne se forme pas ou pratiquement pas de derive acttonyli: en a de I'oxygene. Ceci est en accord a la fois avec la grande difference de labilites entre hydrogenes secondaires en a du carbonyle et hydrogines primaires en n de l'oxygene et avec d'eventuels transferts d'hydrogene.
Le dernier point a souligner est la competition entre I'acetonylation en a (donnant le y-cetoester 4'a) et en /? (donnant le 8-cetoester 4'b) du groupement fonctionnel du butyrate de mtthyle 4 (Schtma 5) ; le phtnomine est comparable a celui observe au cours de l'acetonylation des cyclanones [3].
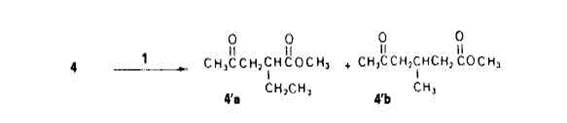
Pour ttudier cette competition, nous avons effectue I'acetonylation avec des solutions de plus en plus dilutes de percarbonate 1 dans le butyrate 4; les pourcentages relatifs de derives ac6tonylt.s 4'a et 4'b obtenus sont consign& dans le Tableau 3.
Tableau 3. Orientation de lhchtonylation du butyrate de mPthyle (4) en fonction de la concentration de 1 duns 4
1/4 en moles initial 1.5 1:20 1.50 1:lOO
![]() 4'a/4'b 64:36 15:25 80:20 9218
4'a/4'b 64:36 15:25 80:20 9218
Ces resultats mettent en evidence l’accroissement siinultane de la selectivitk de l'ace-tonylation en a du carbonyle et de la dilution des solutions de percarbonate. De la mEme maniere que prectdemment, nous pensons que l'explication de ce phenomene se trouve dam l'existence, pour chaque type de radical, d'une competition entre addition et transfert. lnitialement, l'arrachement d'hydrogene par les radicdux t-butoxyle se produit peut-ttre un peu mieux en /l du carbonyle (l'attaque ne doit cependant pas ttre tres selective et, en outre, les radicaux methyle presents dans le milieu arrachent l'hydrogene plut6t en a du carbonyle). Les radicaux en /l, comparables a des radicdux carbon&, ont un certain caractire nucleophile les rendant a la fois peu aptes a s'additionner sur la double liaison du percarbonate et tres capables d'attaquer les sites en a, deficitaires en electrons, des molecules de substrat; ils ont donc tendance a creer, par transfert, des radicaux en a et ceci d'autant mieux que la concentration en substrat est plus elevee. En ce qui concerne les radicaux en a, nous avons deja dit que leur caractere electrophile pouvait expliquer leur «preference» pour l'addition.
Lors de l'itude de l'acetonylation des cyclanones [3], nous avons observe que la substitution est selective, en a du carbonyle, pour un rapport molaire percarbonate/ substrat &gal a 1:20. Comme nous n'avons pas atteint 100% de selectivite mtme avec un rapport de 1:100, nous pensons que l'influence du groupe carboxyle des esters sur la competition ((addition-arrachement)) se fait moins sentir que celle du groupe carbonyle des cyclanones.
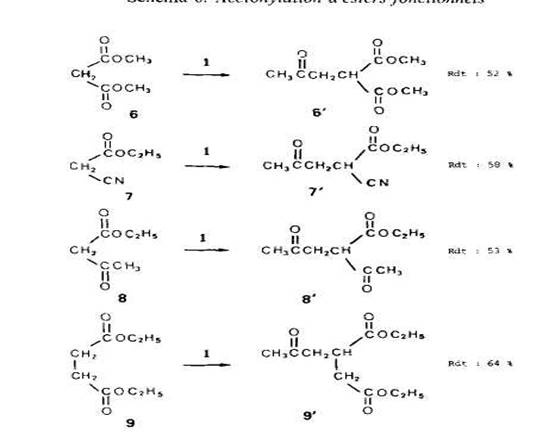
Cas d'esters .fonctionnels. Dans l'etude generale de l'acetonylation [11, nous avons dejja fait etat des resultats obtenus avec le malonate de dimethyle (6) et le succinate de diethyle (9). Nous rappelons ces resultats dans le SchCma 6 en leur ajoutant ceux rela-tifs au cyanacetate d'kthyle (7) et A I'acetylacetate d'ethyle (8).
Les rendements eleves en derives acetonyles (evalues, par rapport i la quantite de 1 mise en jeu, pour des fractions acetonylees purifiees par distillation) sont en accord avec la grande reactivitt des esters fonctionnels en chimie radicalaire [lo].
Tous nos essais ont kte realists avec des rapports molaires initiaux de percarbonate/ substrat de 1:50, c'est-A-dire dans des conditions justifiant que l’on observe selective-ment l'acetonylation en a du (ou d'un) carboxyle. Pour des teneurs en percarbonate suptrieures, nous avons observe la formation de faibles quantites de derives acetonyles correspondant aux parties alcoxyle des molecules ce qui est en accord avec nos observations prkctdentes sur le r6le des transferts d'hydrogene.
En rCssum6, L'etude de l'acetonylation des esters nous a permis de montrer que l'attaque initiale par des radicaux t-butoxyle (et probablement aussi mtthyle) est susceptible de donner des radicaux correspondant aussi bien a la partie alcoxyle qu'a la partie acyloxyle des molecules. Par contre, l'addition sur la double liaison, processus fortement influence par la polarit6 des radicaux [l 11, peut ttre favorisee dans le cas des radicaux en a du carbonyle, forternent electrophiles, et genee dans le cas des radicaux en a de l’oxygene. Ces derniers ont alors la possibilite d'arracher des atomes d'hydrogene en a du carbonyle d'autres molecules pour donner les radicaux correspon-dants. Comme lcs transferts d'hydrogene sont favorises quand on opere avec de grands exces de substrat et que, d'autre part, le meme phtnomene intervient entre les divers sites du cat6 acyloxyle, on conclut qu'aux tres fortes dilutions l'acetonylation tend a se produire selectivement en a du carbonyle. Quand le site en a est un carbone secondaire, on atteint des rendements interessants par rapport au percarbonate 1 mis en jeu.
AcCtonylation des acides. - Les exemples d'additions radicalaires des acides aux alcZnes montrent que le produit majoritaire correspond toujours a l'addition du radical en a du groupe fonctionnel [12]; les principaux produits secondaires proviennent de processus de type ionique [13]. Avec les acides acktique (lo), propionique (ll), butyri-que (12), isobutyrique (13) et dimethyl-3,3 butyrique (14) nous avons aussi observe, par action du percarbonate 1, l'acetonylation sur l'atome de carbone en a du car-boxyle. Les rendements en acides alkyl-2 0x0-Cpentanolques lo', ll', 12', 13' et 14' domes dans le Schha 7 sont ceux de reactions effectutes (a 130°C pendant 2,s h) avec des rapports molaires percarbonate/acide egaux a 1:50 (analyse des fractions acetony-lees aprb esterification par le diazomethane).
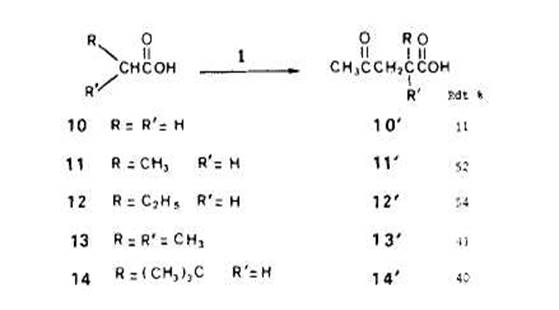
Dans ces conditions, l'acktonylation s'est effectuee selectivement en a du groupe-ment fonctionnel (pour un rapport molaire l/acide butyrique 1:5, les rendements res-pectifs en derivks a et p acCtonylCs sont dans un rapport de 85 A 15). Avec l'acide butyrique, on met encore en evidence l'existence de reactions de transfert entre substrat et radicaux Crees initialement. L'arrachement d'hydrogine par les radicaux t-butoxyle, klectroaccepteurs, porte plut6t sur les sites en fi ou eventuellement y que sur les sites en a dtficitaires en Clectrons. I1 faut donc, qu'au lieu de s'additionner sur la double liaison du percarbonate, les radicaux en f! donnent, par transfert, des radicaux isomires en a pour que les acetonylations apparaissent comme stlectives dans le cas des solutions dilutes de percarbonate.
Dans le cas du butyrate de methyle (4), nous observions encore 8 % d'acetonylation en fi en mettant en jeu un rapport molaire de percarbonate/substrat 1:100. Le fait que, pour I'acide butyrique, la stlectivite soit totale pour un rapport molaire 1:50 montre que I'influence de la fonction carboxyle est plus grande que celle de la fonction carbo-xylate. Ceci est en accord avec les observations [8] [14] sur la ((super r6activitt)) des sites en a d'un carboxyle. D'autre part, d'aprks les rendements d'acetonylation des acides isobutyrique (13) et dimethyl-3,3-butyrique (14) l'encombrement sterique ne di-minue pas de maniere critique la reactivite apparente (avec I'isobutyrate de mkthyle, le rendement en derive acttonyle atteignait a peine 15 YO). I1 faut supposer que la presence du groupe carboxyle en a donne un caractere klectrophile tris prononce aux radicaux correspondants dont la constante de vitesse d'addition sur la double liaison du percarbonate est tr& ClevCe.
Acktonylation des nitriles. - Dans les memes conditions que pour les esters et les acides (13O”C, 2,s h et rapport molaire l/substrat 1:50), nous avons effectue l’acetonylati on de l'acetonitrile (15), du propionitrile (16), du butyronitrile (17) et de I’isobutyronitrile (18) (Schima 8). Ainsi qu'avec les acides, les seuls derives acetonyles obtenus correspondent a l'addition du radical forme en a du groupe fonctionnel sur la double liaison du percarbonate. Comme on peut penser que les radicaux t-butoxyle arrachent initialement plut6t des atomes d'hydrogine en fi, la sklectivite observee ne peut une fois encore s'expliquer qu'en faisant intervenir des transferts d'hydrogine.
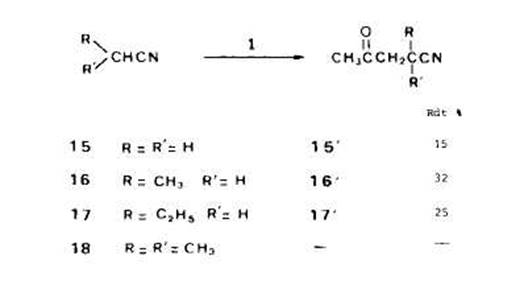
L'impossibilite dans laquelle nous nous sommes trouvts de rkaliser l'acetonylation de l'isobutyronitrile 18 est un peu surprenante, encore que des observations analogues aient deji ete rapportees [lob]. Avec l'acide isobutyrique, en effet, nous avions atteint des rendements en derive acetonyle de I'ordre de 40%. Peut-ttre faut-il penser que le radical tertiaire en a du groupe nitrile est tris fortement stabilisk et ne s'additionne pas sur la double liaison du percarbonate; cette hypothese est d'ailleurs ttayte par le fait que le produit majoritaire avec I'isobutyronitrile est le dehydrodimere correspondant au couplage de deux radicaux tertiaires. Les faibles rendements observes, meme dans le cas du propionitrile et du butyronitrile, sont aussi en accord avec une stabilisation des radicaux en a, stabilisation expliquke par les possibilitirs de dklocalisation offertes par le groupe nitrile superieures a celles qui existent dans le cas des esters ou des acides [15].
Conclusions. ~ L'etude de l'acetonylation d'esters, acides et nitriles confirme que, dans les reactions de type addition radicalaire, les produits formes sont rarement reprt-sentatifs des radicaux libres issus de l'arrachement d'hydrogdne par des radicaux z-bu- toxyle. Cet arrachement ((initial)) peut porter sur des carbones en p (voire y) du groupe fonctionnel ou sur la partie alcoxyle des molecules d'ester mais des transferts d'hydrogene entre les carbones en a du carbonyle et les radicaux ainsi obtenus crtent des radicaux en a qui, s'additionnant sur la double liaison du percarbonate, accroissent les proportions de derives ac6tonylCs en a.
Les transferts d'hydrogene sont favorists si I’on opere avec des solutions tres di-Ides de percarbonate de 0, O-t-butyle et 0-isopropenyle, si bien qu'avec des rapports molaires rkactif/substrat de l'ordre de 1:50 a 1:100, on realise sklectivement I'acetonylation des acides, esters, nitriles (et aussi, rappelons-le, citones) en a du groupe fonctionnel. Comme les rendements sont souvent elevts (plus de 50% en derive acetonyle isole, par rapport au percarbonate mis en jeu), que les produits secondaires (t-butanol, acktone, acttonylacktone et, quelquefois, dthydrodimere du substrat) sont faciles a eliminer par simple distillation et que le substrat en exces peut &tre aisement rkcupertt et rkutilistt, l'acetonylation radicalaire constitue une bonne mithode de syn-thkse de y-ceto-acides, esters ou meme nitriles.
Partie experimentale
Percurbonate de 0,O-t-butyle et 0-isopropPnyle (I). II est prepare suivant une methode classique [I61 d'ob-tention des peresters par rtaction du chloroformiate d'isoproptnylel ) avec l'hydroperoxyde de t-butyle en presence de pyridine et en solution dans le pentane. Le percarbonate liquide, utilisable sans purification, presente, comme le montre l'etude cinetique de sa decomposition [17], une excellente stabilite thermique; sa manipulation ne pose aucun problhe particulier.
Dkcomposition du percurbonate 1 en solution. Une solution de percarbonate (0,02 mol, 3,5 g) dans 1 mol de substrat est introduite dans un autoclave et chauffee A 130” pendant 2,5 h dans une Ctuve thermoregulee. Le fractionnement des melanges riactionnels est realise par distillation; apres elimination des produits secondaires legers (t -butanol, acetone, acetonylacetone) et recuperation du substrat en ex&, les fractions acbtonylbes, sepa-rees du residu (dehydrodimere du substrat) sont analysts par chromatographie en phase vapeur (quelquefois aprk esttrification au diazomethane).
Principales caracteristiques des derives achtonyles obtenus. - Les temperatures d'tbullition (Eb) sont donnees en “C/Torr, celles de fusion (F), non corrigees, en “C. Les spectres de 'H-RMN om ete enregistres pour des solutions a 10% dans CC4 , le tetramethylsilane &ant pris comme reference interne pour la mesure des deplacements chimiques (6 ppm).
0x0-4-pentanoute de mtthyle (2'a) (Ikvulinate de methyle), Eb: 99-100/24; ng: 1,4235 ([18] Eb: 196/760; ng: 1,4233). 'H-RMN: 2,l (s, 3H, CH,CO); 2,3-2,9 (m. 4H, CH,CH2 ); 3,6 (s, 3H, CH3 0).
Actroxy-4-hutanone-2 (2'b), Eb: 78-81/15. Ce produit a tte identifie par comparaison de ses caracteristiques avec celles d'uu khantillon prepare en refbrence par ahtylation (anhydride acetique) [191 de l'hydroxy-4 buta-none-2.
Mkth~l-2-oxo-4-pmtunoate de mPthyle (3'), Eb: 101-103/25; ng: 1,4285 ([20] Eb: 85-87/15; ng: 1,4270). 'H-RMN: 1,l (d, J = 6,6, 3H, CH,-CH); 2,l (s, 3H, CH,O); 2,l-3,2 (m, 3H, CH,CH); 3,7 (s, 3H, CH3 0).
Les chloroformiates vinyliques sont commercialis& par la SuciitP Nationale des Poudres et E.xplosi/i; 12, quai Henri IV; F-75181 Paris-cedex 04.
Etliyl-2-ono-4-pentunoute de mtthyle (4'a), Eb: 81-83/0,8; ng: 1,4294 ([21] Eb: 73/0,2). 'H-RMN: 0,9 (t, J = 7,0, 3H, CH3 CH2 ); l,l-1,7 (m, 2H, CHZ CH,); 2,O (s, 3H, CH,CO); 2,l-3,0 (m, 3H, CHZ CH); 3,6 (3, 3H, CH,O).
MPth~l-3-oxo-5-hexanoute de mtthyle (4'b). I1 a ett identihe, dans les melanges rkactionnels, par SMjCG. SM: 158 (M + ); 127 ((M - CH3 CO)'); 101 ((M - CH,COCH? )+ ); 85 ((M - CH> CO& HJ + ); 43 ((CH3 CO)').
DimPthyl-2,2-ox-o-4-pentanoate de mtthyk (5'), Eb: 5&52/0,6; ng: 1,4331 ([22] Eb: 91,5-92,5/20). 'H-RMN: 1,15 (s. 6H (CH& ); 2,O (s, 3H, CH,CO); 2,6 (s, 2H, CH2 CO); 3,s (s , 3H, CH,O).
Acitonyl malonate de dimith,yle (6'), Eb: 93-95/0,4; ng: 1,4357 ([23] Eb: 135-136/12; ng: 1,4379). 'H-RMN: 2,1 (s, 3H, CH3 CO): 3,O (d, J = 6,0, 2H, CH,CH); 3,3 (d, J = 6,O 1H, CH,CH); 3,6 (s, 6H, 2CH3 0).
C~~uno-2-oxo-4-pentunoate d'tthyle (7'), Eb: 88-91/0,3; ng: 1,4432 ([24] Eb: 69-71/0,1). 'H-RMN: 1,3 (t. J = 6,Y, 3H, CH,CH,); 2,l (s, 3H, CH3 CO); 3,O (d, J = 5,9, 2H, CHZ CO); 3,8 (I, J = 5,9, lH, CHCOO); 4,2 (q, J = 6.9, 2H, CH,O).
AcPtyl-2-oxo-4-pentroute d'gthyle (W ), Eb: 80-8l/0,2; nf : 1.4398 ([25] Eb: 126-128/14; ng: 1,4385). 'H-RMN: 1,2 (1, J = 6,6, 3H, CH,CH,); 2,l et 2.2 (2 s, 6H, 2 CH,CO); 2,9 (d, J = 6,2, 2H, CH,CH); 3,8 (t, J = 6,2, lH, CHCOO); 4,l (9, J = 6,6, 2H, CH,CH,).
AcPtonyl succirzute de ditthvie (Y ), Eb: 80-82/0,2; nf: 1,4418 (1261 Eb: 135/1,3; ng: 1,4400). 'H-RMN (CD,COCD,): 1,2 (1. J = 7.3, 6H, 2CH,CHZ ); 2,l (s, 3H, CH,CO); 2,3-2,9 (m, 5H, CH,CHCH,); 4,O (q, J = 7,3, 4H, 2 CH,O).
Acide 0x0-4-pentunoique (wide Iiuuliqzre) (lo'), Eb: 80-82/0,3; ng: 1,4501 ([27] Eb: 115-118j5; ng: 1,4460). 'H-RMN: 2,O (s, 3H, CH3 CO); 2,5-2,s (m. 4H, CH2 CH2 ); 10,6 (s, IH, OH).
Acide mPthyl-2-oxo-4-pentan[~~que (ll'), Eb: 101-104/0,4; ng: 1,4438 ([27] Eb: 135-136/8; ng: 1,4410). 'H-RMN: 1,15 (d, J = 6,7, 3H, CH3 CH); 2,l (s, 3H, CH,CO); 2,2-3,3 (m, 3H, CH,CH); 11,8 (s, lH, OH).
Acide Pthyl-2-oxo-4-pentano~9ue (lZ'), Eb: 112-115/0,8; ng: 1,4662 ([28] Eb: 132/4; ng: 1,4675). 'H-RMN: 0.9 (t, J = 7,3, 3H, CH3 CH J; 1,3-1,9 (m, 3H, CHZ CH,); 2: l (s, 3H, CH3 CO); 2,l-3,0 (m, 3H, CHZ CHCO); 12,5 (s , IH, OH).
Acide diintthyl, 2. 2-oxo-4-pentunoique (13'), F (hexane, CH2 C12 ): 75 ([29] F (hexane, CH,CI,): 74575). 'H-RMN (CD,COCD,): 1,2 (s, 6H, 2CH3 ); 2,05 (s, 3H, CH,CO); 2,7 (s, 2H, CH2 CO); 12,O (s, lH, OH).
Acide l-hutyI-2-oxo-4-pentano~que (14'), F (hexane, CH,C12 ): 125-126. 'H-RMN (CDCI,): 1,O (s, YH, (CH& C); 2,l (s, 3H, CH3 CO); 2,4-2,9 (m, 3H, CH2 CH); 12,3 (s, lH, OH).
Nous n'avons pas trouvt mention de cet acide dans la litttrature; comme ses bomologues, il a hte identifie par CG apres transformation en ester mkthylique (diazomethane); son analyse centhsimale a donne des rbsultats en accord a 0,3% prks avec les valeurs tbeoriques.
0x0-4-pentunenitrile (Ituulonifrile, 153, Eb: 97/22; ng: 1,4316 ([30] Eb: 95-109/12). 'H-RMN: 2,l (s , 3H, CH,CO); 2,6 (m, 4H, CH2 CH2 ).
MPthyl-2-oxo-4-pentunenitrile (16'), Eb: 105-108/22; ng: 1,4260 ([31] Eb: 68/1, ng: 1,4288). 'H-RMN: 1,3 (d. J = 6,6, 3H, CH,CH); 2,1 (s, 3H, CH3 CO); 2,43,3 (m, 3H, CH2 CH).
Ethyl-2-oxo-4-pentanenitrile (17'), Eb: 95-96/0,8; ng: 1,4349 ([32] Eb: 85-87/0,05). 'H-RMN: 0,9-1,9 (m, 5H, CHj CH2 ); 2,l (F, 3H, CH,CO); 2,2-3,0 (m, 3H, COCHZ CH).