| Скачать .docx |
Реферат: Химическая термодинамика
Московский Авиационный Институт
(Технический Университет)
Кафедра физической химии
Курсовая работа
на тему:
"Химическая термодинамика"
Выполнил: Павлюк Д.В
Проверила: Селиванова С.И.
Содержание:
I. Теоретическая часть
1. Введение……………………………………………………………..3
2. Законы термохимии…………………………………………………3
3. Элементы термодинамики………………………………………….4
4. Первое начало термодинамики…………………………………….5
5. Элементы второго начала термодинамики………………………..9
6. Энтропия…………………………………………………………….11
II. Экспериментальная часть………………………………………16
III. Расчетная часть…………………………………………………17
Список используемой литературы……………………………………19
В результате химической реакции выделяется или поглощается энергия, так как реакция сопровождается перестройкой энергетических уровней атомов или молекул веществ, участвующих в ней, и веществ, образующихся в ходе реакции.
Реакции, при которых наблюдается выделение энергии, называются экзотермическими (Q>0).
Реакции, идущие с поглощением энергии, называются эндотермическими (Q<0). Выделение или поглощение энергии в результате процесса зависит от соотношения количеств энергии, затраченных на разрыв или возбуждение химических связей первоначально взятых веществ, и энергии, выделяющейся в результате образования новых химических связей в продуктах реакции.
Величина энергии отдельной химической связи очень мала. Её удобно выражать в электронвольтах на атом. Поскольку обычно в реакциях участвуют относительно большие количества веществ, то общие количества энергии получаются также большие. Так, элементарный расчет показывает:
на 1 атом: 1эВ=1,6·10-19 Кл∙1В = 1,6. 10-19 Дж,
на 1 моль: 1,6∙10-19 ∙6,02∙1023 =9,65∙104 Дж/моль = 96,5 кДж/моль.
Энергия, образующаяся в результате химических реакций, может выделяться в разных формах, но, конечно, в эквивалентных количествах. Так, например, фотохимические процессы при фотографии развиваются при поглощении квантов лучистой энергии галидами серебра и, наоборот, можно построить источник когерентного излучения—лазер, работающий на энергии химических реакций.
Затрачивая электрическую энергию, можно выделять нужные вещества из растворов или расплавов путем электролиза, с другой стороны, можно получить энергию за счет химических реакций, протекающих в гальванических элементах или аккумуляторах.
Чаще всего в, результате химических реакций выделяется или поглощается тепловая энергия. Поэтому раздел химии, изучающий энергию химических реакций, исторически стал называться термохимией, а изменение энергии называется тепловым эффектом химической реакции и измеряется в килоджоулях на моль образовавшегося или сгоревшего вещества. Так как в зависимости от условий, в которых протекает химическая реакция, возможно выделение или поглощение работы расширения газов (p=const), то различают тепловой эффект реакции при (p=const) Qp и тепловой эффект реакции при (v=const) Qv , хотя разница между ними обычно невелика.
ЗАКОНЫ ТЕРМОХИМИИ
Первый закон термохимии (Лавуазье и Лаплас, 1780—1784):
тепловой эффект образования данного соединения в точности равен, но обратен по знаку тепловому эффекту его разложения.
Из закона Лавуазье—Лапласа следует невозможность построить вечный двигатель I рода, использующий энергию химических реакций.
Второй закон термохимии (Г. И. Гесс, 1840):
тепловой эффект химической реакции не зависит от характера и последовательности отдельных ее стадий и определяется только начальными и конечными продуктами реакции и их физическим состоянием (при p=const или при v=const).
Г. И. Гесс первый принял во внимание физическое состояние реагирующих веществ, так как теплоты изменения агрегатных состояний веществ накладываются на тепловой эффект реакции, увеличивая или уменьшая его.
Утверждение закона Гесса о том, что тепловой эффект процесса не зависит от его отдельных стадий и их последовательности, дает возможность рассчитывать тепловые эффекты реакций для случаев, когда их определить экспериментально или очень трудно, или вообще невозможно.
Применение закона Гесса чрезвычайно расширило возможности термохимии, позволяя производить точные расчеты тепловых эффектов образования целого ряда веществ, опытные данные по которым получить было трудно.
Закон Гесса в наши дни применяют главным образом для расчета термодинамических функций—энтальпий, которые сейчас используются для термохимических расчетов. Термохимия, исторически сложившаяся раньше термодинамики, в настоящее время претерпела некоторые изменения и стала разделом химической термодинамики.
ЭЛЕМЕНТЫ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ
Химическая термодинамика изучает изменения энергии в результате процессов в материальных системах, приводящих к изменению состава и свойств физических тел, из которых построена данная система.
Термодинамической системой называется комплекс взаимодействующих между собой физических тел, мысленно обособленный от окружающей среды.
Системы бывают изолированные, в которых энергообмен и массообмен с окружающей средой отсутствуют, и замкнутые, в которых возможен энергообмен с окружающей средой, но не возможен обмен веществом. Незамкнутые системырассматриваются в термодинамике необратимых процессов.
Системы можно разделить на гомогенные или однородные, не имеющие физических границ раздела между отдельными частями, так как во всех частях системы свойства одинаковы(например, ненасыщенный раствор), и системы гетерогенные, или неоднородные, разделяющиеся на отдельные части физическими границами раздела, на которых свойства системы резко изменяются. Часть гетерогенной системы, ограниченная физическими границами раздела, называется фазой. Например, насыщенный раствор, соприкасающийся с растворяемым веществом, представляет собой гетерогенную систему.
Состояние системы определяется физическими параметрами; в простейшем случае идеального газа — это давление и температура, так какv =f(p. Т).
Изменение параметров системы вызывает процесс. Если процесс заключается в последовательном изменении параметров, приводящих в конечном итоге систему в исходное состояние, то такой процесс называется циклом.
Химическая термодинамика, так же как и общая термодинамика, основана главным образом на двух законах (началах).
ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ
Первое начало термодинамики, окончательно сформулированное Джоулем в середине XIXв., представляет собой закон сохранения энергии. Для замкнутых систем, обменивающихся энергией с окружающей средой, уравнение первого закона термодинамики имеетвид:
![]() (1)
(1)
где Q — энергия, сообщенная системе; ΔU— приращение внутренней энергии системы; А — работа, совершенная системой.
Энергия, сообщенная системе (Q), может быть тепловой или другой формой энергии, так как первый закон термодинамики справедлив для любых процессов. Если система поглощает энергию, то Q принимает положительное значение, т. с. знак Q обратен знаку теплового эффекта реакции:
Q = ¾ Q (2)
Внутренняя энергия системы (U) включает все виды энергии, заключенные в веществах, составляющих систему, кроме энергии, созданной гравитационными, электрическими или магнитными нолями, а также кроме кинетической энергии системы в целом (для движущейся системы). Таким образом, U ¾ сумма всех видов тепловой энергии движения элементарных частиц, энергии связи и энергии агрегатных состояний. Это сложная термодинамическая функция, полностью определяемая состоянием системы или соответствующим сочетанием параметров (р и Т). Если система поглощает энергию, то запас внутренней энергии растет (ΔU>0).
Если работа совершается системой, то А — положительная величина; если же работа совершается над системой, то А отрицательна (например, сжатие газа).
Как Q, так и А в уравнении (1) характеризуют процесс и от состояний системы (начального и конечного) зависят неоднозначно, так как из начального состояния подойти к конечному состоянию можно разными путями и с различным поглощением энергии и различной величиной работы. Поэтому уравнение (1) мы не можем записать в дифференциальной форме, так как только одно приращение ΔU однозначно определяется параметрами состояния р, v, Т.
Если известен закон изменения параметров в данном процессе, то уравнение первого закона термодинамики можно записать в дифференциальной форме и исследовать математически. В области применения химических реакций наиболее часто встречаются процессы, протекающие при постоянном объеме (изохорический) и при постоянном давлении (изобарический).
1. Изохорический процесс: v = const. В этом случае параметры р и Т связаны между собой уравнением Гей-Люссака, р/Т = const . Уравнение (1) записывается в дифференциальной форме:
dQ=dU+dA. (3)
Но если объем постоянен, значит работа расширения или сжатия газа совершаться не может:dA==pdv=0 . Следовательно,dQ = - dU ;
приравниваем частные производные по температуре:
![]()
или
dU = Cv dT, (4)
где Сv — теплоемкость при постоянном объеме. Уравнение (4) позволяет вычислять изменение внутренней энергии системы приизменении температуры, если не происходит каких-либо изменений агрегатного или полиморфного состояния.
Как известно, при химической реакции внутренняя энергия изменяется: если энергия выделяется, то это соответствует уменьшению запаса внутренней энергии, и наоборот. Поэтому тепловой эффект и изменение внутренний энергии имеют обратные знаки:
U = -Qv . (5)
2.Изобарический процесс: р = const . В этом случае по закону Гей-Люссака v/T= const. Кроме того, из уравнения (3) не выпадают отдельные члены, так как при постоянном давлении расширение и сжатие газа возможно, как и нагревание и охлаждение. В этом случае dQ=dU+pdv. После интегрирования в пределах 1—2 получим:
![]()
Выражение в скобках (U + pv) представляет собой термодинамическую функцию, которую назовем энтальпией Н:
H=U+pv . (6)
Энтальпия — это энергосодержание системы, включающее внутреннюю энергию и работу. Тогда
![]() (7)
(7)
Если система поглощает энергиюQ1-2 , то ΔН больше нуля, и если в этой системе происходит химическая реакция, то она будет эндотермической:
![]() (8)
(8)
Так как в дальнейшем мы будем использовать понятие разности энтальпий химической реакции, то необходимо помнить соотношение:
| Экзотермические реакции | Эндотермические реакции |
| ΔH >0; Q p <0 |
Разность энтальпий химической реакций обратно по знаку тепловому эффекту реакции при постоянном давлении. Для вычисления энтальпии исходим из соображений, чтоQ = ΔH ; приравниваем частные производные по температуре:
![]() (9)
(9)
или d(ΔН )=C p dТ , где С р —теплоемкость при постоянном давлении. При расчете ΔН следует учитывать не только изменение энергосодержания системы в зависимости от температуры, но и изменение агрегатных и полиморфных состояний, при котором происходит поглощение энергии при постоянной температуре:
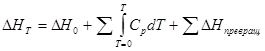 (10)
(10)
Таким образом, энтальпия — сложная математическая функция, определяющая энергию, необходимую для приведения системы в данное состояние, и учитывающая изменение внутренней энергии и совершаемую работу.
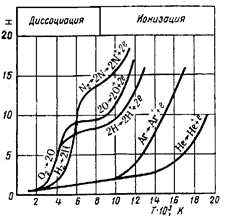 |
На рисунке приведены кривые зависимости энтальпии от температуры для газов, используемых как плазмообразователи в плазмотронах. Для исследования процессов, происходящих в материальных системах, мы пользуемся не абсолютными значениями энтальпий, а их изменением (разностью) между начальным и конечным состояниями системы. Разности энтальпий мы можем измерять с любой степенью точности, отсчитывая энтальпии не от абсолютного |
нуля, а, от любого, но всегда одного и того же уровня. За такой уровень приняты стандартные условия: Т =298,15 К, р =1,013∙105 Па.
Кроме того, для термохимических расчетов приняты следующие два условия:
1. Разность энтальпий простых веществ (ΔН
0
) в состоянии, устойчивом при стандартных условиях, принимается равной нулю. Например:![]() , но
, но ![]() (так как для образования атомарного водорода при стандартных условиях надо затратить энергию диссоциации, равную 217,9 кДж/моль).
(так как для образования атомарного водорода при стандартных условиях надо затратить энергию диссоциации, равную 217,9 кДж/моль).
2. Разность энтальпий сложного вещества обратна по знаку и равна тепловому эффекту при постоянном давлении (![]() ) реакции его образования из простых веществ в состоянии,
устойчивой при стандартных условиях, т.е. энтальпии образования. Например:
) реакции его образования из простых веществ в состоянии,
устойчивой при стандартных условиях, т.е. энтальпии образования. Например: ![]() ¾ 241,8 кДж/моль;
¾ 241,8 кДж/моль; ![]() + 90,37 кДж/моль.
+ 90,37 кДж/моль.
В настоящее время стандартные разности энтальпий (ΔН
0
) и их зависимости от температуры (![]() ) можно найти в справочной литературе для очень большого числа неорганических и органических соединений.
) можно найти в справочной литературе для очень большого числа неорганических и органических соединений.
Термохимические расчеты с использованием табличных данных значительно упростились. Рассмотрим пример расчета разности энтальпий химической реакции в общем виде для уравнения
aA+bB=cC+dD
где А, В, С, D — символы реагирующих веществ: а, Ь, с, d — стехиометрические коэффициенты.
Исходные вещества (аА+ b В ) соответствуют начальному состоянию системы, и сумма их энтальпий вычитается, так как они в результате процесса исчезают, конечные продукты (cC+dD ), составляющие конечную систему, появляются в процессе, и их энтальпии входят со знаком плюс. Если данное вещество в уравнение химической реакции входит с коэффициентом, отличным от единицы, то при суммировании энтальпий эти коэффициенты надо взять как множители.
Во избежание возможных ошибок надо суммирование энтальпий производить непосредственно под уравнением химической реакции
aA+bB=cC+dD
![]()
Подставляя значения энтальпий из справочной литературы, находим ![]() реакции.
реакции.
Чтобы получить разность энтальпий реакций для более высоких температур, чем стандартные, используют зависимость разности энтальпий от температуры и учитывают при этом изменения энергии, потребной для нагрева данных веществ и для изменения ихфазовых состояний:
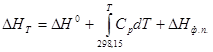 (11)
(11)
Для многих веществ эти функции рассчитаны и приведены в справочных таблицах (ΔНT ¾Н0 ).
Если абсолютное значение разности энтальпий реакций достаточно велико (300—400 кДж), то в первом приближении температурной зависимостью можно пренебречь, так как теплоемкости измеряются в Дж/(моль∙К), а разности энтальпий—в кДж/моль, т.е. на 3 порядка выше.
Для органических соединений в справочных таблицах часто приводится разность энтальпий горения этих веществ, рассчитанная для случая образования жидкой воды, так как обычно определения производятся в калориметрических бомбах, охлаждаемых по окончании опыта до комнатной температуры.
Зная разность энтальпий сгорания, легко определить разностьэнтальпий образования органического вещества. Схема расчета приведена для общего случая горения органического вещества:
![]()
![]()
Отсюда
![]()
Атомы других элементов (Cl, N, S и т.д.), входящие в состав органической молекулы, при горении выделяются в молекулярном виде или в виде устойчивых оксидов (SO2 , P2 O5 ), так как горение происходит в атмосфере кислорода (3∙105 Па).
ЭЛЕМЕНТЫ ВТОРОГО НАЧАЛА ТЕРМОДИНАМИКИ
Первое начало термодинамики — закон сохранения энергии — рассматривает уже свершившиеся процессы, но не указывает направление процесса химической реакции, ее возможность и полноту протекания, а это представляет собой основную задачу при исследовании любого процесса, особенно высокотемпературного.
Так, например, водород и кислород, соединяясь со взрывом, при обычных температурах образуют воду, при высоких температурах реагируют обратимо, а при температуре выше 4000 К существование водяного пара практически невозможно. Таким образом,разность энтальпий реакции еще не определяет возможности ее протекания в данных конкретных физических условиях.
Изменение химической энергии зависит от условий, поэтому развитие химических реакций, как и всех остальных процессов, например тепловых, определяется вторым началом термодинамики. Согласно второму началу термодинамики (сформулированному в окончательной форме Клаузиусом и Гельмгольцем в середине XIX в.) теплота может переходить в работу только при наличии разности температур и не целиком, а с определенным термическим коэффициентом полезного действия (η):
![]() (12)
(12)
где A — работа, полученная за счет перехода теплоты от тела с высокой температурой (Т 1 ) к телу с низкой температурой (Т 2 );Q 1 — теплота, взятая у нагретого тела с температуройТ 1 ;Q 2 — теплота, отданная холодному телу с температуройТ 2 .
Учитывая, что температура выражена в абсолютной шкале, мы видим, что КПД тепловых машин вообще невелик. Например, КПД теплоэлектроцентрали, работающей с перегревом пара до 673 Кис конденсатором при Т 2 =323 К
![]() или 52%
или 52%
(И это без учета всех остальных потерь в рабочем цикле турбин и механических потерь!)
Таким образом, для любых процессов, протекающих под действием разности потенциалов (grad P
), каковой для тепловых процессов является разность температур, для электрических — разность потенциалов, для механических — разность высот и т.д., общим является сравнительно низкий коэффициент полезного действия. Значение КПД обращается в единицу, если в уравнении (12) Т
2
![]() 0, но абсолютный нуль недостижим. Следовательно, всю энергию нагретого тела при температуре Т
1
, в работу превратить нельзя.
0, но абсолютный нуль недостижим. Следовательно, всю энергию нагретого тела при температуре Т
1
, в работу превратить нельзя.
Заряд q проходит разность потенциалов, совершая работу
A=q(U1 -U2 ). (13)
Однако всю энергию он отдает только в том случае, если U2 →O.
Вода вращает турбину при перепаде уровней воды: верхний бьеф — нижний бьеф плотины:
![]() (14)
(14)
Однако всю энергию положения (потенциальную) вода отдаст только в том случае, если h2 → 0, т. е. вода будет падать до центра земли, что невозможно.
Таким образом, при совершении работы часть общей энергии системы остается неиспользованной.
При течении химических реакций энтальпия начальных продуктов не может вся перейти в работу или теплоту, так как в конечных продуктах реакции сумма энтальпий не равна нулю. Если градиент движущих сил (Т, U , h и т. д.) равен нулю, то и работа, совершающаяся в процессе, равна нулю, а система будет находиться в состоянии равновесия: при Т1 =Т 2 закончится теплообмен: электрический заряд не осуществляет работы, если U1 = U2 турбины не работают при спущенной плотине; химическая реакция будет достигать равновесия, когда количество полученных конечных продуктов равно количеству разложившихся конечных продуктов на первоначальные за единицу времени.
Исследуя выражение для КПД тепловой машины, Клаузиус ввел новую термодинамическую функцию, которую назвал энтропией . В самом деле:
![]() или
или ![]()
![]()
отсюда
![]() или
или ![]() (15)
(15)
Таким образом, при проведении цикла в идеальной тепловой машине (цикл Карно) и получении механической работы отношение полученной теплоты к температуре нагретого источника равно такому же отношению для холодного источника. Так как Q является в уравнении (15) приращением энергии, то можно это отношение записать в дифференциальной форме для элементарных циклов:
![]()
суммируя изменения по всему циклу тепловой машины, можно записать
![]() (16)
(16)
где dQ — приращение теплоты; Т — соответствующая температура;![]() — интеграл по замкнутому контуру.
— интеграл по замкнутому контуру.
Подынтегральное выражение Клаузиус принял за приращение новой функции S — энтропии:
![]() или
или ![]() (17)
(17)
Энтропия представляет собой функцию параметров состояния (р, v, Т ) и может оценить направление процесса в системе, стремящейся к равновесию, так как для идеального или равновесного процесса ее изменение равно нулю:dS =0.
В самом деле, заменяя dQ на изменение внутренней энергии и работыdQ=dU+pdv , можно записать
![]() (18)
(18)
ЕслиU=const и v = const , то в идеальном процессеdS=0 , что, по существу, определяет равновесие системы (обратимый процесс), и в этом случае энтропия стремится к максимальному значению:
S→Smax .
Приращение энтропии определяется развитием необратимых процессов, протекающих самопроизвольно, которые прекращаются только при достижении равновесия в системе.
Однако требование постоянства внутренней энергии системы исключает возможность использования только одной этой функции для исследования химических реакций, при которых внутренняя энергия веществ, составляющих систему, неизбежно меняется.
Гиббспредложил другую термодинамическую функцию, исследуя которую можно определить направление процессов в системе, стремящейся к равновесию приT=const иp=const :
G=H ¾ TS (19)
гдеG — энергия Гиббса (или термодинамический потенциал, как назвал эту функцию Гиббс); Н —энтальпия; S —энтропия; Т — абсолютная температура.
Опуская все математические исследования термодинамической функцииG , можно считать, что функция G для системы, стремящейся к равновесию, убывает, при достижении равновесия она принимает минимальное значение (G→Gmin ), а ее приращение обращается в нуль (ΔG=0 ).
ЭНТРОПИЯ
Наиболее информативной термодинамической функцией в уравнении (19) является энтропияS .
Значение энтропии легко определить только для состояния идеального газа. Используем для вычисления S
уравнение (18), где dU
— изменение внутренней энергии, равное для идеального газа С
v
dT
т.е. теплоемкости при постоянном объеме, умноженной на приращение температуры:pdv
— приращение работы, которое можно представить как![]() , заменив р
наRT/v
. Отсюда
, заменив р
наRT/v
. Отсюда
![]() (20)
(20)
После интегрирования в пределах 0 ¾ T получаем
![]() (21)
(21)
рис. 2 Схема для расчета энтропии при самопроизвольном смешивании двух газов. |
гдеST — энтропия при температуре Т ;S0 — энтропийная постоянная; С v — теплоемкость при постоянном объеме; v — молярный объем. Таким образом, энтропия моля идеального газа является функцией Т и р (так как молярный объем зависит от Т и р ). Выражение (21) применимо лишь для чистого идеального газа, так как для смесей газов, даже при отсутствии между ними химических реакций, энтропия смеси будет возрастать за счет необратимых процессов диффузии, приводящей к распределению компонентов по всему объему газовой смеси. Рассмотрим процесс самопроизвольного смешения двух газов. Пусть в двух частях объема, разделенного перегородкой r (рис. 2, а), находится n1 молей первого газа и n2 молей второго газа при р, Т= const . |
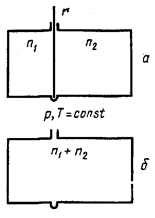
Общая энтропия системы
![]() (22)
(22)
гдеS1
иS
2
— молярные энтропии первого и второго газов. Удалим перегородку r и дадим возможность газам образовать смесь, равномерно распределенную по всему объему ![]() (рис 2,б), гдеv
—молярный объем газа при данных р
и Т
. На каждый моль компонентов смеси приходятся пропорциональные части объема:
(рис 2,б), гдеv
—молярный объем газа при данных р
и Т
. На каждый моль компонентов смеси приходятся пропорциональные части объема:
![]() и
и ![]()
Подставляем значения этих объемов в уравнение (21) и получаем значения энтропии для одного моля компонента в смеси:
![]()
![]()
Общий запас энтропии в смеси газов тоже увеличился:
![]() (23)
(23)
Приращение энтропии в газовой смеси зависит от соотношения чисел молей компонентов n1 и n2 . Если положить, что n1 → 0 , то энтропия этого газа
![]()
но доля, вносимая этим компонентом в общий запас энтропии системы, также стремится к нулю: ![]() . В то же время если n1
→ 0
, то (n1
+n2
)/n2
стремится к 1, а
. В то же время если n1
→ 0
, то (n1
+n2
)/n2
стремится к 1, а ![]() тоже стремится к нулю. Следовательно, при исчезновении одного из компонентов газовой смеси энтропия другого компонента станет равна энтропии чистого газа:
тоже стремится к нулю. Следовательно, при исчезновении одного из компонентов газовой смеси энтропия другого компонента станет равна энтропии чистого газа:
![]() .
.
Так как отношение (n1 +n2 )/n1 представляет собой величину, обратную молярной доле данного компонента
![]()
то в общем виде можно записать выражение энтропии моля газа в смеси следующим образом:
![]() (24)
(24)
![]() (25)
(25)
Полученное выражение определяет очень важное понятие, а именно—рассеяние вещества, так как если N → 0 , то энтропия стремится к ∞.
В природе существуют так называемые рассеянные элементы, общее содержание которых, вообще говоря, не так мало, но они присутствуют в очень малых количествах в различных минералах, водах и т. д. Для того чтобы выделить эти элементы в свободном виде, их сначала надо концентрировать, а это трудно и требует очень большой затраты энергии. Так, например, морская вода содержит ничтожные количества золота, но так как воды в мировом океане очень много, то и золота в ней тоже огромное количество» Однако если золото выделять из морской воды известными методами, то оно будет «дороже золота».
Приращение энтропии при смещении газов —RlnNi можно использовать при рассмотрении любых разбавленных растворов. В растворах более концентрированных взаимодействие между молекулами растворенного вещества уменьшает их активность, и поэтому в таких случаях вместо величин концентрации в уравнение под' ставляют величины «активности» а :
![]()
где а — активность; γ — коэффициент активности, стремящийся в разбавленных растворах к единице; Ni , — молярная доля. Энтропия реальных веществ, способных менять свое агрегатное или полиморфное состояние, определяется сложнее, так как для каждого состояния значение энтропии будет иное.
Изменение энтропииΔS при любом превращении вещества можно определить по уравнению
![]() (26)
(26)
где ΔHпревращ — изменение энтальпии при превращении; Тпревращ — температура превращения.
Зависимость энтропии от температуры определяется из уравнения
![]() (27)
(27)
где Ср
— теплоемкость при постоянном давлении![]() . Общая формула температурной зависимости с учетом возможных агрегатных превращений будет
. Общая формула температурной зависимости с учетом возможных агрегатных превращений будет
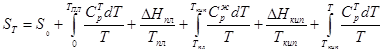 (28)
(28)
Для удобства расчетов и построения таблиц в справочниках приняты стандартные значения энтропии при Т =298,15 К и р = 1,013∙105 Па, т.е. значения при тех же условиях, что и в случае расчета энтальпий. Некоторые значения стандартных энтропии приведены в табл.1 .
Таблица 1. Значения стандартных энтропий S0 для некоторых веществ.
| Вещество | S0 | Вещество | S0 | Вещество | S0 | Вещество | S0 |
| H2 O (г) | 188,74 | H (г) | 114,6 | Cl2 (г) | 223,0 | CO2 (г) | 213,6 |
| H2 O (ж) | 69,96 | H2 (г) | 130,6 | HCl (г) | 186,7 | FeO (кр) | 58,79 |
| H2 O (кр) | 39,33 | O2 (г) | 205,03 | CO (г) | 197,4 | α – Fe (кр) | 25,15 |
Как видно из табл. 1, для воды наблюдается рост энтропии при изменении ее агрегатных состояний от кристаллов к газу.
При переходе вещества от упорядоченного состояния (кристалл) в жидкое или газообразное состояние энтропия моля вещества растет.
Больцман, развивая статистические идеи в термодинамике, впервые показал сущность энтропии для идеальных газов, определив ее пропорциональность термодинамической вероятностиWi
![]()
Термодинамическая вероятность Wi рассматривается как число возможных способов построения данной системы или число микросостояний, с помощью которых осуществляется данное макросостояние вещества. Естественно, упорядочена система, например кристалл, тем меньше возможных микросостояний (отклонений от равновесного состояния) и тем меньше энтропия.
II. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ.
ОПРЕДЕЛЕНИЕ ТЕПЛОВОГО ЭФФЕКТА НЕЙТРАЛИЗАЦИИ СЕРНОЙ КИСЛОТЫ
Цель работы - определение теплового эффекта реакции нейтрализации и проверка закона Гесса.
Нейтрализация 1 г-экв любой сильной кислоты сильным основанием в достаточно разбавленном растворе сопровождается почти одинаковым экзотермическим тепловым эффектом, отвечающим одному и тому же процессу ¾ образованию 1 моля жидкой воды из гидратированных ионов ![]() и
и ![]() по уравнению:
по уравнению:
![]() ΔНнейтр
= ¾ 55.9 кДж/г-экв
ΔНнейтр
= ¾ 55.9 кДж/г-экв
Оборудование 1. Внутренний стакан калориметра(1). 2. Калориметр (2). 3. Мешалка(3). 4. Термометр(4). 5. Бюретка(5). Рассчитываем тепловой эффект реакции нейтрализации одного г/экв кислоты (ΔНнейтр
) no формуле (Здесь Q¾количество теплоты, выделившейся в калориметре;n¾ количество г/экв кислоты в 200 мл 0,4н раствора). Значение Q вычисляем во формуле |
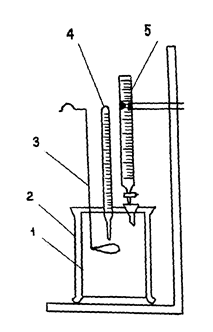 |
ственно массы стакана, мешалки, кислоты и щелочи; Сст ¾ удельная теплоемкость стакана (стекла), равная 0,69 кДж/кг∙К;
СМ - удельная теплоемкость мешалки (стали),равная 0,42 кДж/кг∙К; Ск , Сщ ¾ удельные теплоемкости кислоты и щелочи (4.2 кДж/∙К).
РАСЧЕТЫ.
| Масса внешнего стакана калориметра | Масса мешалки | Температура, С˚ | ||||
| Начальная темп., Т1 | Конечная темп., Т2 | Разность , ΔТ=Т2 -Т1 | ||||
| 132 гр. | 23.2 гр. | 22.8 | 27.6 | 4.8 | ||
| 132 гр. | 23.2 гр. | 22.9 | 25 | 26 | 2.1 | 3.1 |
Уравнение реакции:
![]()
Нормальность ¾ это количество эквивалентов в 1 л.
![]()
49гр. ¾ 1н.
X гр. ¾ 2н.
X = 98 г. / экв.
1.
Q = (0.69 ∙ 132 + 0.42 ∙ 23.2 + 4.2 ∙ 200 + 4.2 ∙ 40) ∙ 4.8 = 5322.35
Δ Н нейтр = ¾ Q / N = 5322.35 /98 = -54.3 кДж
Ошибка опыта:
![]()
2.
Q1 = (0.69 ∙ 132 + 0.42 ∙ 23.2 + 4.2 ∙ 200 + 4.2 ∙ 40) ∙2.1=2328.5
Q2 = (0.69 ∙ 132 + 0.42 ∙ 23.2 + 4.2 ∙ 200 + 4.2 ∙ 40) ∙3.1=3437.35
Q общ = Q1 + Q2
Δ Н нейтр = ¾ Q общ / N = 5765.85 /98 = -58.8 кДж
IV. РАСЧЕТНАЯ ЧАСТЬ.
Вариант № 5
№ 1
![]()
![]()
![]()
Ответ: ΔН = ¾ 204.8 кДж.
№ 2
Дано:
![]() ;
; ![]()
![]() ;
; ![]()
![]() ;
; ![]()
![]()
![]()
![]()
![]()
Что бы пошла реакция прямо, необходимо, что бы Δ G <0 :
![]()
Δ H-TΔS<0
T> Δ H/ΔS
T>2.8
Ответ: реакция пойдет при Т
>
298+2.8
![]() Т
>
300.8 К
Т
>
300.8 К
№3
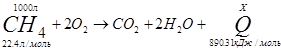
![]()
Δ H= ¾ ΔQ
![]()
Ответ: 39745 Дж
№4
Дано:
N2 ¾ 1 моль
H2 ¾ 3 моль
![]()
![]()
![]()
А)
![]()
![]()
Общая энтропия системы: ![]() + 3 ∙
+ 3 ∙ ![]() = 583.3
= 583.3
В)
![]()
Энтропия при данной реакции будет:
![]()
№5
4.9гр. Х гр.
![]()
1 моль 2 моль
98 г. 112г.
![]() ЭКОН
=56/1=56
ЭКОН
=56/1=56
2Nкон =1л.
56/2=28 гр.
28гр ¾ 1л. ![]()
5.6 ¾ Х л.
Ответ: 200 мл.
Список используемой литературы:
1. Учебное пособие к лабораторному практикуму по химии, М., изд-во МАИ, 1984г.
2. Ахметов Н.С Общая и неорганическая химия, М., Высшая школа, 1980г.
3. В.В. Фролов Химия М., Высшая школа, 1986г.