| Скачать .docx |
Курсовая работа: Синтез и идентификация сульфата магния MgSO4
Федеральное агентство по образованию
государственное образовательное учреждение
высшего профессионального образования
Поморский государственный университет им. М.В.Ломоносова
Естественно-географический факультет
Курсовая работа по неорганической и аналитической химии
Синтез и идентификация сульфата магния
MgSO 4
Выполнила студентка 3 курса
11 группы отделения химии
Васюк Кристина
Станиславовна
к.х.н., доцент Попова Людмила
к.п.н., доцент Швакова Элла
Валерьевна
2009
Введение........................................................................................................................................ 3
Глава 1. Обзор литературы....................................................................................................... 7
1.1. Характеристика и химические свойства сульфата магния......................................... 7
1.2. Способы получения сульфата магния.............................................................................. 8
1.3. Техника безопасности при работе в химической лаборатории.................................... 9
1. Основные правила работы в лаборатории общей химии.............................................. 9
2. О пожарах в лаборатории и способах их устранения.................................................. 11
3. Медицинская помощь в лаборатории............................................................................ 11
4. Техника безопасности при работе с сульфатом магния............................................... 12
1.4. Качественный анализ MgSO4 и его возможных примесей........................................... 14
1.5. Количественное определение сульфата магния и его примесей.................................. 28
1.6. Количественный анализ:................................................................................................. 29
1. Приготовление установочного вещества..................................................................... 29
2. Приготовление рабочего раствора................................................................................. 30
3. Приготовление аммонийного буфера........................................................................... 30
4. Стандартизация рабочего раствора................................................................................ 31
5. Приготовление анализируемого раствора.................................................................... 32
6. Определение Mg2+ методом прямой комплексонометрии, титрант – ЭДТА .......... 33
7. Катионообменная хроматография ................................................................................. 34
8. Ионообменная хроматография....................................................................................... 35
9. Определение SO4 2- обратным комплексонометрическим титрованием ................... 36
Глава 2. Химический эксперимент........................................................................................ 39
2.1. Задачи эксперимента...................................................................................................... 39
2.2. Синтез сульфата магния............................................................................................... 39
2.2.2. Расчет синтеза............................................................................................................ 40
2.2.3. Подготовка к синтезу................................................................................................ 41
2.2.4. Техника безопасности при работе с серной кислотой ........................................... 42
2.2.5. Синтез вещества......................................................................................................... 44
Анализ синтезированного соединения и возможных примесей.......................................... 46
1. Оборудование и реактивы.............................................................................................. 46
2. Приготовление и стандартизация растворов для комплексонометрического определения Mg2+ и SO4 2- ....................................................................................................................................... 48
Качественный анализ сульфата магния и возможных примесей:..................................... 51
1. Количественное определение Mg2+.......................................................................... 53
2. Количественное определение SO4 2- ......................................................................... 55
Итоговая таблица:................................................................................................................. 58
Заключение................................................................................................................................. 59
Библиографический список..................................................................................................... 60
Приложение 1………………………………………………………………………………… 61
Приложение 2 ……………………...…………………………………………………….……61
Схема …………………………………………………………………………………………..62
Введение.
Курсовая работа является важным способом контроля и этапом в изучении любой дисциплины. Здесь в полной мере проявляются профессиональные знания и качества, которыми обладает студент.
Данная курсовая работа посвящена синтезу и анализу сульфата магния.
Сульфат магния широко применяется в различных отраслях промышленности [11]:
1. В химической промышленности он применяется в технологических процессах получения H2 SO4 и MgO; при производстве синтетических моющих средств.
2. В сельском хозяйстве сульфат магния используется в качестве удобрения почвы под сельскохозяйственные культуры.
3. В текстильной промышленности сульфат магния применяют в технологических процессах обработки тканей; для приготовления огнестойких составов для пропитки различных материалов; используется как наполнитель материалов, утяжелитель шелка и хлопка, протрава для покраски тканей и как отбеливающий компонент.
4. В медицине в качестве успокаивающего и спазмолитического средства; применение сульфата магния в комплексной терапии больных дискулярной энцефалопии позволяет уменьшить последствия ишемического повреждения головного мозга и купировать общемозговые нарушения.
5. В фармацевтической промышленности сульфат магния применяется в процессах изготовления антибиотиков.
6. Строительная индустрия:
1). Сульфат магния применяется как добавка для устройства дорожных и аэродромных оснований и покрытий;
2). Входит в состав магнезиального цемента ( это композиция из каустического магнезита (продукт обжига MgСО3 при температурах до 700 0 С в форме MgО) и солей магния, главным образом MgСl2 и MgSO4 . Водные растворы последних часто называют «затвердителями». Без затвердителей каустический магнезит, смешанный с водой, твердеет медленно.
7. В целлюлозно-бумажной промышленности сульфат магния используется как наполнитель, а также как компонент позволяющий сохранить и улучшить физико-механические показатели бумаги при использовании отбеливателей (особенно хлорсодержащих) и для получения огнестойких изделий из бумаги.
Цели исследования в данной работе:
1. Синтезировать и идентифицировать соединение;
2. подтвердить его чистоту
3. провести количественный и качественный анализ.
Задачи исследования:
По литературным источникам:
- составить характеристику исследуемого вещества
- осуществить подбор и обработку методик синтеза и анализа;
- составить схему качественного анализа
- описать способы приготовления растворов, их стандартизацию.
В ходе эксперимента:
- синтезировать вещество;
- определить выход препарата;
- определить качественный и количественный состав синтезируемого соединения.
Глава 1. Обзор литературы
1.1. Характеристика и химические свойства сульфата магния
Сульфат магния (магний сернокислый, горькая соль), MgSO4 - это белый кристаллический порошок с плотностью 2,66 г/см3 . При 1127°С разлагается с выделением SO2 . Хорошо растворяется в воде (слабый гидролиз по катиону). Образует кристаллогидраты: MgSO4 *H2 O (бесцветные кристаллы с моноклинной решеткой с плотностью 2,6 г/см3 ), MgSO4 *6H2 O (кристаллы с моноклинной решеткой с плотностью 1,70 г/см3 ) и MgSO4 *7H2 O (бесцветные кристаллы с ромбической решеткой с плотностью 1,68 г/см3 ).
Сульфат магния используют как сырье для получения MgO, в производстве магнезиальных цементов, в качестве утяжелителей хлопка и шелка, протравы при крашении тканей, как наполнитель бумаги, спазмолитическое, слабительное и желчегонное лекарственное средство [11].
Химические свойства:
1. Сульфат магния хорошо растворяется в воде, гидролизуется по катиону:
MgSO4 (разб.) + 4H2 O → [Mg(H2 O)4 ]2+ + SO4 2- ,
[Mg(H2 O)4 ]2+ + H2 O → [Mg(H2 O)3 (OH)]+ + H3 O+ ;
2. Вступает в реакции обмена:
MgSO4 + BaCl2 → BaSO4 ↓ + MgCl2
3. Может взаимодействовать с концентрированной серной кислотой:
MgSO4 + H2 SO4 → Mg(HSO4 )2
4. Реагирует с щелочами:
MgSO4 + NaOHразб → Mg(OH)2 ↓ + Na2 SO4
5. Взаимодействует с концентрированным аммиаком:
MgSO4 +2 NH4 OHконц → Mg(OH)2 ↓ + (NH4 )2 SO4
1.2. Способы получения сульфата магния.
По Ключникову Н. Г. [6]
По данной методе, сульфат магния можно получить, растворяя оксид магния в горячей серной кислоте.
MgO + H2 SO4 → MgSO4 + H2 O
По методике, описанной в практикуме Корякина и Ангелова [1], MgSO4 можно получить, растворяя карбонат магния в серной кислоте:
MgCO3 + H2 SO4 → MgSO4 + CO2 ↑+ H2 O
Вгорячую30%-ную серную кислоту вносят при энергичном перемешивании MgCO3 (технический) до прекращения вспенивания жидкости. Отфильтрованная проба жидкости не должна давать красного окрашивания с NH4 SCN, в противном случае прибавляют еще MgCO3 . Раствор фильтруют и фильтрат оставляют кристаллизоваться на холоду. На следующий день, выпавшие кристаллы отсасывают на воронке Бюхнера, промывают небольшими количеством ледяной воды и перекристаллизовывают из воды (на 100 г соли берут 40 мл воды).
1.3. Техника безопасности при работе в химической лаборатории
1. Основные правила работы в лаборатории общей химии
1) К работе в лаборатории могут быть допущены лица, прошедшие инструктаж по технике безопасности.
2) Все опыты, реакции и все связанные с ними операции следует делать аккуратно, точно придерживаясь указаний, имеющихся в учебниках, руководствах или сообщённых преподавателем.
3) Лабораторный рабочий стол следует содержать в порядке и чистоте, потому что загрязнения со стола могут попасть в анализируемый раствор и испортить анализ.
4) Лабораторный стол нельзя загромождать посудой и приборами, т.к. все это мешает работе. На столе должно быть только то, что необходимо в данное время для выполнения опыта.
5) Многие работы в химической лаборатории связаны с возможностью загрязнения, а также порчи одежды при попадании на неё кислот и других реактивов, поэтому каждый работающий в химической лаборатории должен надевать халат.
6) Химическая посуда в большинстве случаев тонкостенная и хрупкая, поэтому при небрежном отношении к ней можно её разбить и порезаться. Посуду и приборы следует держать в руках осторожно, не сжимая сильно пальцами. Химическую посуду нельзя резко ставить на стол, особенно, если он покрыт керамикой.
7) Нюхать вещества нужно, не наклоняясь над сосудом и не вдыхая полной грудью, а только направляя к себе пары движением руки.
8) Категорически запрещается пробовать что-либо на язык. Запрещается набирать ртом при помощи пипетки пли трубки ядовитые или едкие жидкости; для этого следует пользоваться грушей.
9) Во избежание случайных отравлений запрещается пользоваться для питья какой-либо химической посудой.
10) Строго запрещается принимать в лаборатории пищу.
11) Все опыты с ядовитыми, вредными для здоровья и дурно пахнущими веществами следует проводить обязательно под тягой.
12) При приготовлении растворов кислот, надо вливать кислоту в воду, а не наоборот, и пользоваться при этом толстостенной или фарфоровой посудой.
13) Производя химические опыты, приборы следует располагать таким образом, чтобы реагирующие вещества в случае разбрызгивания не могли попасть на самого экспериментатора или находящихся вблизи других лиц.
14) Взвешивать твёрдую щелочь нужно обязательно в какой-нибудь стеклянной или фарфоровой посуде.
15) Химические реактивы брать только шпателем, пинцетом или ложечкой (не руками!).
16) Неизрасходованные реактивы не высыпать и не выливать обратно в те сосуды, откуда они были взяты.
17) Жидкости переливать через химические воронки. Склянку, из которой переливают жидкость, необходимо держать этикеткой к руке во избежание её порчи.
18) При нагревании растворов и веществ в пробирке необходимо использовать держатель. Отверстие пробирки должно быть направлено в сторону от себя и других работающих.
19) При неосторожной работе возможны ожоги нагретой стеклянной посудой, тиглями и т.п., поэтому нельзя брать горячую посуду незащищёнными руками.
20) Если на сосуде нет этикетки или надписи и не известно, какой реактив содержится в нём, то пользоваться этим реактивом нельзя.
21) После работы в лаборатории нужно тщательно вымыть руки, так как значительная часть веществ, с которыми приходится соприкасаться в лаборатории, ядовита.
2. О пожарах в лаборатории и способах их устранения
1) Если загорелись деревянные предметы, пожар можно тушить водой, песком и с помощью огнетушителя.
2) Если горит нерастворимое в воде вещество (например, бензин, скипидар, масло и др.), то нельзя применять для тушения воду, потому что пожар не только не будет ликвидирован, но даже может усилиться. Многие огнеопасные органические вещества легче воды и при соприкосновении с нею образуют горящую плёнку.
3) Нерастворимые в воде органические вещества следует тушить песком или накрыванием шерстяным одеялом. Нужно именно накрывать очаг пожара, а не набрасывать, чтобы горящие брызги не разлетались в стороны.
4) В легко доступном месте лаборатории должен висеть проверенный огнетушитель, а также должен быть ящик с песком или листовым асбестом. Работающим в лаборатории необходимо ознакомиться с инструкцией по обращению с огнетушителем, обычно написанной на каждом из них.
5) В случае загорания проводов и электроприборов, находящихся под током, необходимо немедленно выключить ток и тушить огонь сухим углекислотным огнетушителем, сухим песком, покрывалом из асбеста.
6) Если загорелась одежда, следует сначала погасить пламя, накинув на пострадавшего шерстяное одеяло.
3. Медицинская помощь в лаборатории
1) В случае пореза стеклом нужно вначале осмотреть ранку и извлечь из нее осколки стекла, если они есть, а затем обмыть пораненное место, смазать йодом и завязать бинтом или залепить лейкопластырем.
2) При тепловом ожоге надо смочить обожжённое место раствором перманганата калия, затем смазать мазью от ожогов, и перевязать бинтом.
3) При ожоге кислотами поражённый участок кожи быстро промыть большим количеством воды, затем обмыть 2%-ным раствором соды NaНСО3 , смазать обожженное место вазелином и перевязать бинтом.
4) При ожогах щёлочью: смыть водой, а затем 2%-ным раствором уксусной или борной кислоты, после этого смазать борным вазелином или 5%-ным раствором марганцевокислого калия и перевязать бинтом. При ожогах глаз промыть 1%-ным раствором борной кислоты и закапать 1 -2 капли касторового масла.
5) При поражении электричеством выключить ток пли устранить контакт при помощи резиновых перчаток или сухой деревянной палки. [9, 10]
4. Техника безопасности при работе с сульфатом магния.[12]
Магний сернокислый 7-водный пожаро- и взрывобезопасен, по степени воздействия на организм относится к веществам 4-го класса опасности.
Магний сернокислый 7-водный упаковывают в полипропиленовые, полиэтиленовые, бумажные мешки, мягкие специализированные контейнеры разового использования, пакеты с внутренним полимерным покрытием, из полимерных и комбинированных водонепроницаемых материалов или другую влагонепроницаемую тару.
Опасность для человека . Пыль продукта оказывает раздражающее действие на слизистые оболочки глаз и дыхательных путей. При длительном воздействии на организм может вызвать кожные заболевания.
Средства индивидуальной защиты :
1). Защитные очки, респираторы "Кама" или У-2К, хлопчатобумажные, резиновые или поливинилхлоридные перчатки, спецодежда.
2). Одежда должна подвергаться стирке в мыльно-содовом растворе не реже 1 раза в неделю, резиновые перчатки и очки ежедневно обмывать водой, респираторы менять по мере необходимости.
3). Перед едой необходимо тщательно вымыть руки и лицо с мылом, прополоскать рот.
Необходимые действия в аварийных ситуациях:
При утечке, разливе и россыпи
-
просыпанный продукт тщательно собрать сухим способом и использовать в переработке в начале производства.
Освободившуюся тару сжечь или утилизировать в отведенных местах.
При пожаре
-
вызвать пожарную бригаду.
Рекомендуемые средства тушения – вода, пенные огнетушители и песок.
Меры первой помощи
-
при попадании сульфат магния на кожные покровы – промыть загрязненное место водой с мылом.
При попадании в глаза
– немедленно промыть большим количеством воды в течение 15 минут, при необходимости обратиться к врачу.
При вдыхании
– вывести пострадавшего на свежий воздух.
При отравлении
– вызвать рвоту, дать выпить пострадавшему большое количество молока или воды с измельченным активированным углем (5-6 таблеток на стакан воды), обратиться к врачу или доставить пострадавшего в медицинское учреждение, по возможности предъявить тарную этикетку или инструкцию по применению.
1.4. Анализ MgSO4 и его возможных примесей.
Качественный анализ:
Анализ проводится согласно схемы 1.
1. Небольшое количество сухой соли растворить в воде при перемешивании. Если полного растворения не произошло, отделить осадок 1 центрифугированием. Получаем раствор 1 , содержащий Fe 2+ ; Ca 2+ ; Mg 2+ ; Mn 2+ ; Pb 2+ ; Zn 2+ ; NO 3 - ; SO 4 - ; Cl - , и осадок 1 , содержащий CaSO 4 ↓, Ca 3 ( PO 4 )2 ↓, CaCO 3 ↓, PbSO 4 ↓, Pb 3 ( PO 4 )2 ↓, PbCO 3 ↓, PbCl 2 ↓, Mg 3 ( PO 4 )2 ↓, MgCO 3 ↓, Mn 3 ( PO 4 )2 ↓, MnCO 3 ↓, Fe 3 ( PO 4 )2 ↓, FeCO 3 ↓, Zn 3 ( PO 4 )3 ↓, ZnCO 3 ↓ [3].
2. Анализ раствора 1
2.1. Осаждение Ca2+ и Pb 2+ из раствора 1 [3]:
Дробно. К части раствора 1 прибавить смесь 2N H2 SO4 +C2 H5 OH, центрифугированием отделить осадок 3 от раствора 3 .
Са 2+ + Н 2 SO4 = CaSO4 ↓ + 2H+
Pb2+ + Н 2 SO4 = PbSO4 ↓ + 2H+
Получаем раствор 2, содержащий Fe 2+ , Mg 2+ , Zn 2+ , Mn 2+ , Cl - , NO 3 - , SO 4 - и осадок 2, содержащий CaSO 4 ↓ и PbSO 4 ↓.
2.2. Открытие Pb 2+ [3]:
К осадку 2 добавить (NH4 )2 SO4, немного нагреть. Осадок 3 PbSO4 ↓ отделить от раствора 3 , содержащего Ca2+ центрифугированием:
Са SO4 ↓+(NH4 )2 SO4 .→ (NH4 )2 [ Са (SO4 )2 ]
2.2.1. Осадок 3 растворить, добавив 30% раствор ацетата аммония, смесь нагреть;
PbSO4 ↓ + 4CH3 COONH4 = (NH4 )2 [Pb(CH3 COO)4 ] + (NH4 )2 SO4
Получим раствор 4, содержащий Pb2+ .
2.2.2. К 2 каплям раствора 4 , содержащего Pb2+ , добавить 2 капли 2NCH3 COOH и 1 каплю раствора KI. Затем добавить несколько капель дистиллированной воды и 2N CH3 COOH, смесь нагреть до растворения осадка (если осадок не растворился добавить еще кислоты), затем медленно или быстро охладить. В присутствии Pb2+ сначала образуется желтый осадок, который при нагревании растворяется, а при охлаждении образуется вновь, но уже в виде золотистых чешуйчатых кристаллов:
Pb 2+ + 2 KI = PbI 2 ↓+ 2 K +
PbI 2 ↓ + 2 KI = K 2 [ PbI 4 ]
PbI 2 ↓ + 2 CH 3 COOH = ( CH 3 COO )2 Pb + 2 HI
2.3. Открытие Ca 2+ из раствора 3, полученного в 2.2. [3]:
Микрокристаллоскопически. На предметное стекло нанести каплю раствора 3 и добавить к нему каплю серной кислоты, смесь перемешать и рассмотреть под микроскопом. Образуется белый осадок сульфата кальция (гипса), кристаллы которого под микроскопом имеет форму отдельных игл или игл собранных в пучок:
SO4 2- + Ca2+ = CaSO4 ↓

а) б)
Кристаллы Ca2 SO4 *2H2 O, образующиеся в разбавленных (а) и в концентрированных (б) растворах кислот
3. Анализ раствора 2.
3.1. Осаждение Fe 2+ , Mg 2+ , Mn 2+ , Ca 2+ , Pb 2+ из раствора 2 [3] :
К 2 мл раствора 2 добавить поташ до прекращения образования осадка. Смесь слегка нагреть. Осадок 4 , содержащий FeCO 3 ↓, MnCO 3 ↓, MgCO 3 ↓, PbCO 3 ↓, CaCO 3 ↓, ZnCO 3 ↓ отделить от раствора 5 , содержащего Cl - , NO 3 - , SO 4 2- :
Pb2+ +K2 CO3 = PbCO3 ↓+2K+
Ca2+ +K2 CO3 = CaCO3 ↓+2K+
Fe2+ +K2 CO3 = FeCO3 ↓+2K+
Mn2+ +K2 CO3 = MnCO3 ↓+2K+
Mg2+ +K2 CO3 = MgCO3 ↓+2K+
Zn2+ +K2 CO3 = ZnCO3 ↓+2K+
3.2. Открытие Cl - , NO 3 - , SO 4 - из раствора 5 [3]:
Дробно. Проверить раствор 2 на полноту осаждения катионов тяжелых металлов, добавив к части раствора поташ. Если осадок образуется, то удаление катионов необходимо повторить. К раствору 2 прибавить раствор уксусной кислоты до нейтральной реакции среды - это раствор 3.
K2 CO3 +2 CH3 COOH = H2 О +CO2 ↑+2CH3 COO К
3.2.1. Открытие Cl - [3]:
Дробно. К 3 каплям раствора 5 добавить по 3 капли 2NH2 SO4 и KMnO4 . к отверстию в пробирки, не касаясь стенок, поднести влажную йодокрахмальную бумажку. Если газ выделяется плохо, смесь нагреть:
Йодокрахмальная бумажка синеет:
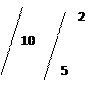 10
KCl
+ 2
KMnO
4
+ 8
H
2
SO
4
= 5
Cl
2
+ 2
MnSO
4
+ 6
K
2
SO
4
+ 8
H
2
O
10
KCl
+ 2
KMnO
4
+ 8
H
2
SO
4
= 5
Cl
2
+ 2
MnSO
4
+ 6
K
2
SO
4
+ 8
H
2
O
Окисление: MnO 4 - +8 H + + 5 e = Mn 2+ + 4 H 2 O
Восстановление: 2 Cl - -2 e = Cl 2
_____________________________________________
2 MnO 4 - + 16 H + + 10 Cl - = 2 Mn 2+ + 8 H 2 O + 5 Cl 2
Йодокрахмальная бумажка синеет вследствие:
Cl - + 2 I - = I 2 + 2 Cl -
I 2 + крахмал = синий комплекс
3.2.2 Открытие SO 4 - [3]:
Микрокристаллоскопия. На предметное стекло нанести каплю раствора 5 и добавить к нему каплю хлорида кальция, смесь перемешать и рассмотреть под микроскопом. Образуется белый осадок сульфата кальция (гипса), кристаллы которого под микроскопом имеет форму отдельных игл или игл собранных в пучок:26
SO4 2- + Ca2+ = CaSO4 ↓


а) б)
Кристаллы Ca2 SO4 *2H2 O, образующиеся в разбавленных (а) и в концентрированных (б) растворах кислот
3.2.3. Открытие NO 3 - [3]:
В пробирку с 3 каплями раствора 5 прибавить 6 капель концентрированной серной кислоты и небольшой кусочек медной проволоки. Если реакция идет плохо смесь можно слегка подогреть. Медь в присутствии серной кислоты восстанавливает нитрат- ионы до оксида азота (IV) или до бурого газа NO2 ↑:
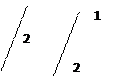 2NaNO3
+ Cu + 2H2
SO4
= CuSO4
+ 2NO2
↑+ Na2
SO4
+ 2H2
O
2NaNO3
+ Cu + 2H2
SO4
= CuSO4
+ 2NO2
↑+ Na2
SO4
+ 2H2
O
Восстановлении : Cu0 – 2e = Cu2+
Окисление : NO3 - + 2H+ + 1e = NO2 ↑+ H2 O
_______________________________________________________________________
Cu0 + 2NO3 - + 4H+ = Cu2+ + 2NO2 ↑+ 2H2 O
4. Анализ осадка 4.
4.1.Перевод тяжелых металлов из осадка 4 в раствор 6 [3]:
Осадок 4 растворить в 2N HNO3 :
FeCO3 ↓+2HNO3 = Fe(NO3 )2 +2H2 O+ CO2 ↑
MnCO3 ↓+2HNO3 = Mn (NO3 ) +2H2 O + CO2 ↑
MgCO3 ↓+2HNO3 = Mg (NO3 ) +2H2 O +CO2 ↑
CaCO3 ↓+2HNO3 = Ca(NO3 )2 +2H2 O+ CO2 ↑
PbCO3 ↓+2HNO3 = Pb(NO3 )2 +2H2 O+ CO2 ↑
ZnCO3 ↓+2HNO3 = Zn(NO3 )2 +2H2 O+ CO2 ↑
Получаем раствор 6 .
4.2.Осаждение Ca 2+ и Pb 2+ из раствора 6 в осадок 2 [3] :
К раствору 6 приливаем раствор K2 SO4 .
Ca(NO3 )2 + K2 SO4 = CaSO4 ↓ + 2KNO3
Pb(NO3 )2 + K2 SO4 = PbSO4 ↓ + 2KNO3
Выпавший осадок CaSO 4 ↓ и PbSO 4 ↓ отделить от раствора 6 и анализировать как в пункте 2.2 – 2.3.
4.2. Определение Fe 2+ [3] :
К раствору 6 прибавим раствор гексоцианоферрата (III) калия. В присутствии Fe2+ выпадает синий осадок комплексного соединения «турнбуленова синь», нерастворимый в кислотах, но разрушающийся в щелочах до гидроксида железа (II), при избытке реагента осадок приобретает зелёный оттенок:
Fe2+ +K3 [Fe(CN)6 ] = KFe[Fe(CN)6 ] ↓+2K+ ,
KFe[Fe(CN)6 ] ↓+2KOH = K3 [Fe(CN)6 ] +Fe(OH)2 ↓.
4.3. Открытие Mn 2+ , Mg 2+ [3] :
В пробирку с раствором 6 добавить избыток 2NNaOH. Образовавшийся осадок 5 , содержащий Mg ( OH )2 ↓, Fe ( OH )2 ↓, Mn ( OH )2 ↓ отделить от раствора 12 , содержащего Zn2+ :
Mg2+ + 2NaOH = Mg(OH)2 ↓ + 2Na+
Fe2+ + 3NaOH = Fe(OH)2 ↓ + 3Na+
Mn2+ + 2NaOH = Mn(OH)2 ↓ + 2Na+
Zn2+ + 2NaOH = Na2 [Zn(OH)2 ]
Разделим получившийся осадок 5 на две равные части 5.1 и 5.2 . В каждой части будет находиться Mg ( OH )2 ↓ , Fe ( OH )2 ↓, иMn ( OH )2 ↓.
4.3.1. Открытие Mn 2+ из осадка 5.2 [3] :
К полученному осадку 5.2. прилить немного серной кислоты. Получим раствор 9 , содержащий Mn 2+ , Mg 2+ , Fe 2+ , Zn 2+ .
Mn(OH)2 ↓ + H2 SO4 = Mn SO4 + 2H+
Mg(OH)2 ↓ + H2 SO4 = Mg SO4 + 2H+
Fe(OH)2 ↓ + H2 SO4 = Fe SO4 + 2H+
Zn(OH)2 ↓ + H2 SO4 = Zn SO4 + 2H+
В пробирку перенести несколько кристаллов персульфата аммония, прибавить 3-5 капель 2N азотной кислоты и 1-2 капли 1%-ого раствора нитрата серебра. Смесь нагреть до 70˚ С. В нагретую смесь внести палочкой одну каплю исследуемого раствора. Смесь можно подогреть. При наличии в растворе Mn 2+ раствор станет розовым или малиновым. При большой концентрации ионов Mn 2+ в раствормогут высыпать бурые хлопья MnO ( OH )2 . В этом случае опыт повторить, предварительно разбавив анализируемый раствор дистиллированной водой в 2-5 раз.
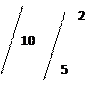 2MnSO4
+ 5(NH4
)2
S2
O8
+8H2
O = 2HMnO4
+ 5(NH4
)2
SO4
+ 7H2
SO4
2MnSO4
+ 5(NH4
)2
S2
O8
+8H2
O = 2HMnO4
+ 5(NH4
)2
SO4
+ 7H2
SO4
Окисление: Mn2+ + 4H2 O +5e = MnO4 - + 8 H+
Восстановление: S2 O8 2- -2e = 2SO4 2 -
2Mn2+ + 8H2 O+ 5 S2 O8 2- = 2 MnO4 - + 16H+ + 10 SO4 2-
4.3.2. Открытие Mg 2+ из осадка 5.1 [3]:
К образовавшемуся осадку 5.1. добавить 5 капель пероксида водорода, смесь нагревать 2-3 минуты:
 1. 2Fe(OH)2
↓ +4 NaOH + H2
O2
= 2Fe(OH)3
↓ + Na+
1. 2Fe(OH)2
↓ +4 NaOH + H2
O2
= 2Fe(OH)3
↓ + Na+
Окисление: Fe2+ + 3OH- - 1e = Fe(OH)3 ↓
Восстановление: H2 O2 + 2e = 2OH-
Fe(OH)2 ↓+ 4OH - + H2 O2 = Fe(OH)3 ↓+ 2OH-
 2. Mn(OH)2
↓ +2 NaOH + H2
O2
= MnO(OH)2
↓ +2 Na+
+ H2
O
2. Mn(OH)2
↓ +2 NaOH + H2
O2
= MnO(OH)2
↓ +2 Na+
+ H2
O
Окисление: Mn2+ +4OH - - 2e = MnO(OH)2 + H2 O
Восстановление: H2 O2 + 2e = 2OH-
Mn(OH)2 ↓ +2OH - + H2 O2 = MnO(OH)2 ↓ + H2 O
Полученный осадок 6, содержащий Mg ( OH )2 ↓, MnO ( OH )2 ↓ и Fe ( OH )3 ↓, после охлаждения отцентрифугировать; центрифугат отбросить, а осадок растворить в насыщенном растворе хлорида аммония:
Mg ( OH )2 ↓ + 2 NH 4 Cl = MgCl 2 + 2 NH 3 * H 2 O
Полученный раствор 8 отделить от осадка 7 центрифугированием, осадок 7 отбросить. К полученному раствору 8 прибавить гидрофосфат натрия, добавить аммонийный буфер (рН 8-10). В присутствии Mg2+ выпадает белый кристаллический осадок, кристаллы которого под микроскопом имеют форму шестилучевых звезд или снежинок.
Mg 2+ + NH 3 * H 2 O + Na 2 HPO 4 = ( NH 4 ) MgPO 4 ↓ + 2 Na + + H 2 O
Рисунок:
5. Осаждение тяжелых металлов [3]:
К оставшейся части раствора 1 прибавить раствор K 2 CO 3, после завершения реакции раствор отделить от осадка центрифугированием. Проверить раствор на полноту осаждения тяжелых металлов K 2 CO 3 .
Получаем раствор 5 (анализ раствора см 3.2. открытие Cl - , NO 3 - , SO 4 - из раствора 5: ) и осадок 9
Fe2+ +K2 CO3 = FeCO3 ↓+2K+
Mn2+ +K2 CO3 = MnCO3 ↓+2K+
Pb2+ +K2 CO3 = PbCO3 ↓+2K+
Mg2+ +K2 CO3 = MgCO3 ↓+2K+
Ca2+ +K2 CO3 = CaCO3 ↓+2K+
Zn2+ +K2 CO3 = ZnCO3 ↓+2K+
5.1. перевод тяжелых металлов из осадка 9 в раствор 10 [3] :
Осадок 9 растворить в 2N азотной кислоте:
FeCO3 ↓+2HNO3 = Fe(NO3 )2 +2H2 O + CO2 ↑
MnCO3 ↓+2HNO3 = Mn (NO3 )2 +2H2 O + CO2 ↑
MgCO3 ↓+2HNO3 = Mg (NO3 ) +2H2 O + CO2 ↑
CaCO3 ↓+2HNO3 = Ca(NO3 )2 +2H2 O + CO2 ↑
PbCO3 ↓+2HNO3 = Pb(NO3 )2 +2H2 O+ CO2 ↑
ZnCO3 ↓+2HNO3 = Zn(NO3 )2 +2H2 O+ CO2 ↑
Получаем раствор 10.
5.2.Осаждение Ca2+ и Pb 2+ из раствора 10 [3] :
К раствору 10 прибавить смесь 2N H2 SO4 +C2 H5 OH, центрифугированием отделить осадок от раствора.
Са 2+ + Н 2 SO4 = CaSO4 ↓ + 2H+
Pb2+ + Н 2 SO4 = PbSO4 ↓ + 2H+
Получаем раствор 11 и осадок 2
Анализ осадка 2 смотреть в пункте 2 (без осаждения) – 2.3.
6. Анализ раствора 11
6.1. дробное открытие Fe 2+ [3] :
См 4.1.
6.2. открытие Zn 2+ [3] :
К раствору в пробирке добавить избыток щелочи NaOH.
Mg2+ + 2NaOH = Mg(OH)2 ↓+ 2Na+
Mn2+ +2 NaOH = Mn(OH)2 ↓+ 2Na+
Fe2+ + 2NaOH = Fe(OH)2 ↓+ 2Na+
Zn2+ + 2NaOH = Na2 [Zn(OH)4 ]
Получим осадок 5, содержащий Mg ( OH )2 ↓, Mn ( OH )2 ↓, Fe ( OH )2 ↓, и раствор 12, в состав которого входит Na 2 [ Zn ( OH )4 ], которые разделим центрифугированием. К полученному раствору 12 добавить несколько капель дитизона C13 H12 N4 S зеленого цвета и содержимое пробирки сильно встряхнуть. При наличие в растворе ионов цинка капля органического растворителя окрасится в малиновый цвет, а водный раствор приобретет красную или розовую окраску. Если окрасится только органический слой, то это не говорит о наличии в растворе ионов цинка.
Zn2+ + 2NaOH = Zn(OH)2 + 2Na+
Zn(OH)2 + 2NaOH = Na2 [Zn(OH)4 ]
Zn 2+ + 4 OH - = [ Zn ( OH )4 ]2-
Анализ осадка 5 смотреть в пункте 4.3.
7. Анализ осадка 1
7.1. Открытие CO 3 2- [3] :
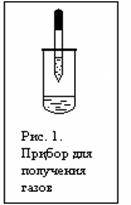
Дробно. Собрать прибор, как это показано на рисунке 1. В пипетку набрать известковую воду. В пробирку поместить намного осадка 1 . К осадку добавить 5 капель 2N раствора HNO3 . Пробирку быстро закрыть пробкой с вставленной в нее пипеткой:
FeCO 3 ↓ + 2 HNO 3 = Fe ( NO 3 )2 + CO 2 ↑+ H 2 O
PbCO3 ↓ + 2HNO3 = Pb(NO3 )2 + CO2 ↑+ H2 O
CaCO3 ↓ + 2HNO3 = Ca(NO3 )2 + CO2 ↑+ H2 O
 MgCO3
↓ + 2HNO3
= Mg(NO3
)2
+ CO2
↑+ H2
O
MgCO3
↓ + 2HNO3
= Mg(NO3
)2
+ CO2
↑+ H2
O
MnCO3 ↓ + 2HNO3 = Mn(NO3 )2 + CO2 ↑+ H2 O
ZnCO3 ↓ + 2HNO3 = Zn(NO3 )2 + CO2 ↑+ H2 O
Известковая вода в пипетке мутнеет:
Ca(OH)2 + CO2 ↑ = CaCO3 ↓ + H2 O
7.2. Перевод Pb 2+ и Cl - в раствор [3] :
К осадку 1 добавить воды и немного нагреть. Раствор 13 отделить от осадка 11 центрифугированием:
PbCl 2 ↓+ 4 H 2 O = [ Pb ( H 2 O )4 ] Cl 2
7.3. открытие Pb 2+ [3] :
См. п.2.2.
7.4. открытие Cl - [3] :
К оставшейся части раствора 13 добавить поташ. Получим осадок 14 и раствор 17 , которые отделим центрифугированием:
Pb 2+ + K 2 CO 3 = PbCO 3 ↓+2 K +
Нейтрализация раствора 17:
K 2 CO 3 +2 CH 3 COOH = 2 CH 3 COOK + CO 2 ↑ + H 2 O
Открытие Cl- смотреть в пункте 3.2.1.
8. Анализ осадка 11
8.1. Открытие Pb 2+ и SO 4 2- [3] :
К осадку 11 добавить 30% раствор ацетата аммония, смесь нагреть;
PbSO4 ↓ + 4CH3 COONH4 = (NH4 )2 [Pb(CH3 COO)4 ] + (NH4 )2 SO4
Получим раствор 14 , содержащий Pb2+ , SO4 2- .
8.1.1. Открытие Pb 2+ [3] :
См 7.3.
8.1.2. Открытие SO 4 2- [3] :
К раствору 14 прилить K2 CO3 . Полученный раствор 16 отделить от осадка 13 центрифугированием. Осадок 13 отбросить.
Pb 2+ + K 2 CO 3 = PbCO 3 ↓+2 K +
Нейтрализация раствора 16:
K 2 CO 3 +2 CH 3 COOH = 2 CH 3 COOK + CO 2 ↑ + H 2 O
Анализ раствора 16 см 3.2.2 .
8.2. Перевод осадка 12 в раствор15 [3] :
К осадку 12 добавить раствор 2NHNO3 .
FeCO3 +2 HNO3 = Fe(NO3 )2 + CO2 + 2H2 O
Fe3 (PO4 )2 + 6HNO3 = 3Fe(NO3 )2 + 2H3 PO4
MnCO3 ↓+ 2HNO3 = Mn (NO3 )2 + 2H2 O + CO2 ↑
Mn3 (PO4 )2 + 6HNO3 = 3Mn(NO3 )2 + 2H3 PO4
MgCO3 ↓+ 2HNO3 = Mg (NO3 ) + 2H2 O + CO2 ↑
Mg3 (PO4 )2 + 6HNO3 = 3Mg(NO3 )2 + 2H3 PO4
CaCO3 ↓+ 2HNO3 = Ca(NO3 )2 + 2H2 O + CO2 ↑
Ca3 (PO4 )2 + 6HNO3 = 3Ca(NO3 )2 + 2H3 PO4
ZnCO3 ↓+ 2HNO3 = Zn(NO3 )2 + 2H2 O + CO2 ↑
Zn3 (PO4 )2 + 6HNO3 = 3Zn(NO3 )2 + 2H3 PO4
8.3. Открытие PO 4 3- [3] :
К раствору 15 добавить Na2 CO3 .
Fe2+ + N а 2 CO3 = FeCO3 ↓+2 Na+
Mn2+ + Na2 CO3 = MnCO3 ↓+2 Na+
Pb2+ + Na2 CO3 = PbCO3 ↓+2Na+
Mg2+ + Na2 CO3 = MgCO3 ↓+2Na+
Ca2+ + Na2 CO3 = CaCO3 ↓+2Na+
Zn2+ + Na2 CO3 = ZnCO3 ↓+2Na+
Получим осадок 9 и раствор 18 , содержащий PO4 3- .
8.3.1. Нейтрализация раствора 18 [3] :
К раствору 18 прибавить раствор уксусной кислоты до нейтральной реакции среды:
Na 2 CO 3 +2 CH 3 COOH = H 2 О+ CO 2 ↑+2 CH 3 COONa
К полученному нейтральному раствору добавить несколько капель молибденовой жидкости. Образовавшийся желтый кристаллический осадок додекамолибдофосфата свидетельствует о наличие в растворе фосфат-ионов:
PO4 3- + 12(NH4 )2 MoO4 + 24HNO3 ! = (NH4 )3 [PMo12 O40 ] ↓ + 12H2 O + NH4 NO3 + +3NO3 -
8.4. Анализ осадка 9 [3] : См 5.1.
| ион | Метод | Вид титрования | Титрант | Индикатор | рН | |||
| SO4 2- | 1. Комплексонометрия 2. Ионообменная хроматография, катионит в Н – форме. |
Обратное | титрант 1: BaCl2 титрант 2: ЭДТА |
Хромоген черный | 8-10 (аммиачный буфер) | |||
| Fe2+ | 1. Перманганатометрия, 2. ОВТ, |
Прямое | KMnO4 | Безындика-торный | 4-6 (сернокислая среда) | |||
| Са2+ | Комплексонометрия | Прямое | ЭДТА | Мурексид | 12 (щелочь) | |||
| Pb2+ | Комплексонометрия | Прямое | ЭДТА | Ксиленоловый оранжевый | 5 (ацетатный буфер) | |||
| Cl- | 1. Осадительное, 2. Аргентометрия, 3. метод Мора |
Прямое | AgNO3 | K2 CrO4 | 7 | |||
| NO3 - | 1. Катионообменная хроматография, катионит в Н-форме 2. КОТ, алкалиметрия |
Прямое | NaOH | ф/ф | 7 | |||
| CO3 2- | КОТ, алкалиметрия | обратное | титрант 1: HCl титрант 2: NaOH | ф/ф | 7 | |||
| Mn2+ | 1. ОВТ, 2. Перманганатометрия |
Прямое | KMnO4 | Безындика-торный | 7 (ZnO) |
|||
| Mg2+ | Комплексонометрия | Прямое | ЭДТА | Эриохром | 9,5-10 (аммонийный буфер) |
|||
1.5. Количественное определение сульфата магния и его примесей [4]:
Таблица 1
1.6. Количественный анализ:
Количественное определение Mg 2+ проводится прямым комплексонометрическим титрованием.
Количественное определение SO 4 2- проводится обратным комплексонометрическим титрованием.
1. Приготовление установочного вещества [4]:
Приготовить 50 мл 0,1N раствора MgSO4 *7H2 O.
1.1 Расчет навески:
1. fэ = ½
2. Мэ = fэ*М =1/2*246,47=123,235 г
3. g(MgSO4 ∙7H2 O)=Мэ *V*CN =123,235 г/моль*0,05 л*0,1 моль/л = 0,6162 г
1.2 Взятие навески (сначала на ТВ, а потом на АВ) [4].
mпуст.кап =
mкап.с в-вом =
mкап.с ост. =
gпракт. =
1.3 Растворение навески [4]:
1.3.1. Заполнить мерную колбу на 1/3 объема дистиллированной водой.
1.3.2. Высыпать навеску в мерную колбу через сухую воронку. Смыть небольшим количеством воды остатки навески с воронки и горлышка колбы.
1.3.3. Растворить соль. Если вещество растворяется плохо, колбу слегка нагреть на плитке до полного растворения вещества.
1.3.4. Охладить колбу до комнатной температуры. Довести объем раствора в колбе дистиллированной водой до метки по нижней границе мениска (последнюю каплю воды добавлять глазной пипеткой).
1.3.5. Закрыть колбу пробкой и перемешать 15-20 раз. Содержимое колбы вылить в чистую посуду.
1.4 Рассчеты:
1. Т(MgSO4 ∙7H2 O)= g(MgSO4 ∙7H2 O)/V(колбы)
2. СN (MgSO4 ∙7H2 O)=Т(MgSO4 ∙7H2 O)*1000/Мэ(MgSO4 ∙7H2 O)
3. К=gпракт ./gтеор.
2. Приготовление рабочего раствора [4]:
Приготовить 50 мл 0,1N раствора ЭДТА.
2.1. Расчет навески:
1. fэ = ½
2. Мэ(ЭДТА) = fэ*М=½*372,24=186,12 г/моль
3. gтеор. (ЭДТА) = Мэ *V*CN = 186,12 г/моль*0,05 л*0,1 моль/л=0,9306 г≈0,93 г
2.2. Взятие навески (на ТВ).
2.3. Растворение навески: проводить как в п. 1.3.
3. Приготовление аммонийного буфера:
1) Колбу на 10 мл заполняем на ⅓ дистиллированной водой.
2) Добавляем туда 57 мл 25%-ного раствора NH3 *H2 O и 6,7 г NH4 Cl.
3) Перемешиваем и доводим объем до метки дистиллированной водой.
4) Приготовленный раствор переливаем в чистую посуду. Среду проверяем рН-метром (8-10).
4. Стандартизация рабочего раствора [4]:
MgSO4 + H2 Ind ═ H2 SO4 + MgInd
![]()
![]() HOOCH2
C CH2
COONa
HOOCH2
C CH2
COONa
![]()
![]() MgInd + N─ CH2
─ CH2
─ N → H2
Ind . NaOOC H2
C CH2
COOH
MgInd + N─ CH2
─ CH2
─ N → H2
Ind . NaOOC H2
C CH2
COOH
![]() -
OOC─H2
C CH3
COONa
-
OOC─H2
C CH3
COONa
+ N─ CH2 ─ CH2 ─ N
![]() Na OOC ─H2
C CH3
COO-
Na OOC ─H2
C CH3
COO-
![]()
![]()
![]()
![]() Mg
Mg
Таблица 2
| № | Vсоли | VЭДТА | Условия титрования |
1 2 3 |
2 2 2 |
V1 V2 V3 Vср |
1. Прямое, индикаторное титрование. 2.Титрант – ЭДТА. 3.В колбе для титрования 2 мл MgSO4 ∙7H2 O + 2 мл аммонийного буфера + сухой эриохром. 4. рН=10. 5. Титровать энергично перемешивая от винно-красной до синей окраски. Объем избыточной капли не вычитать. |
Стандартизация раствора ЭДТА по сульфату магния
Расчеты:
1. CN (ЭДТА)= CN (MgSO4 ∙7H2 O)*V(MgSO4 ∙7H2 O)/V(ЭДТА)
2. Т(ЭДТА)= CN (ЭДТА)*Мэ(ЭДТА)/1000
3. Т(ЭДТА/ MgSO4 ∙7H2 O)= CN (ЭДТА)*Мэ(MgSO4 ∙7H2 O)/1000
5. Приготовление анализируемого раствора:
Приготовить 50 мл 0,1N раствора MgSO4
5.1 Расчет навески:
1. fэ =1/2
2. Мэ = 120,36 * ½=60,18г/моль
3. gт =CN *Vл *Mэ =0,1моль/л*0,050л*60,18г/моль=0,3009г
5.2. Взятие навески (сначала на ТВ, затем на АВ):
mпуст.кап =
mкап.с в-вом =
mкап.с ост. =
gпракт. =
5.3. Растворение навески см п. 1.3
6. Определение Mg2+ методом прямой комплексонометрии, титрант – ЭДТА [4]
Таблица 3
Титрование анализируемого раствора
| № | Vсоли | VЭДТА | Условия титрования |
1 2 3 |
2 2 2 |
V1 V2 V3 Vср |
1. Прямое, индикаторное титрование. 2.Титрант – ЭДТА. 3.В колбе для титрования 2 мл MgSO4 + 2 мл аммонийного буфера + сухой эриохром. 4. рН=10. 5. Титровать энергично перемешивая от винно-красной до синей окраски. Объем избыточной капли не вычитать. |
Расчеты:
1. CN (Mg2+ ) = CN (ЭДТА)*V(ЭДТА)/Vопр
2. Т(ЭДТА/Mg2+ ) = CN (ЭДТА)*Мэ(Mg2+ )/1000
3. Т(ЭДТА/ MgSO4 ) = CN (ЭДТА)*Мэ(MgSO4 )/1000
4.ω%пр (Mg2+ ) = [T(ЭДТА/ Mg2+ )*V(ЭДТА) * Vразв * 100] / [gпр. ( MgSO4 )*Vоп ]
5. ω%теор (Mg2+ ) = М(Mg2+ )/М(MgSO4 )=24,305/120,36=0,2019 или 20,19%
6. Dабс = ωпр (Mg2+ )-ωтеор (Mg2+ )
7. Dотн. = Dабс *100% / ωтеор. ( Mg2+ )
8. ω%пр (MgSO4 ) = [T(ЭДТА/ MgSO4 ) *V(ЭДТА) *Vразв *100]/[gпр. (MgSO4 )*Vоп ]
9. Dабс = ωпр (MgSO4 )-ωтеор (MgSO4 )
10. Dотн. = Dабс *100% / ωтеор. ( MgSO4 )
7. Катионообменная хроматография [5]
Катионообменная хроматография необходима для удаления ионов магния, которые мешали бы открытию сульфат-ионов.
7.1. Подготовка катионита к работе [5]:
1 . Взять бюретку с краном объемом 25 мл и диаметром 1 см, в нижнюю часть ее помещаем опорный ватный тампон. В химическом стакане 5 г катионита марки КУ – 2 залить пятикратным объемом дистиллированной воды на 30 минут. Этой взвесью аккуратно заполнить бюретку, лишнюю воду слить через кран. Слой катионита должен быть 10 см, над уровнем катионита должно оставаться 0,5 мл воды, а между частицами катионита должен отсутствовать воздух.
2 . Для перевода катионита в H-форму через колонку пропустить 50 мл 4М HCl, установив краном скорость вытекания, равную 1 капле в секунду.
Приготовление 40 мл 4N раствора HCl:
f=1
Mэ = 36,4612 г/моль
gт =CN *Vл *Mэ = 4 моль/л*0,050 мл*36,461 г/моль=7,2922 г≈7,29 г
3. Взятие навески [4]:
Кислоты, особенно концентрированные, нельзя взвешивать на весах, поэтому нужно пересчитать навеску на объем концентрированной кислоты Т% =С% *ρ/100, где ρ определяется практически по ареометру, С% - по справочнику:
V(HCl конц.) = gтеор (HCl)/ Т% (HCl)
4. Растворение аликвоты [4]:
1) Заполнить мерную колбу на ½ объема дистиллированной водой.
2) Внести туда мерной пипеткой необходимое количество концентрированной кислоты.
3) Перемешать.
4) Довести объем до метки дистиллированной водой, последние капли добавляя глазной пипеткой, перемешать 15-20 раз, перелить раствор в чистую посуду.
8. Ионообменная хроматография[5]:
2R-H+ + MgSO4 = R2 -Mg2+ + H2 SO4
Взять мерной пипеткой 10 мл анализируемого раствора и пропустить через колонку с катионитом в Н-форме, поддерживая скорость 2 мл в минуту. Вытекающий из колонки элюат собрать в коническую колбу. Промыть колонку дистиллированной водой (~60-80 мл), пропуская ее небольшими порциями. Новую порцию воды приливать тогда, когда жидкость в колонке почти достигнет поверхности анионита. Сделать проверку последних порций элюата на полноту омывки (индикатор не должен менять свой цвет). Промывные воды и основной элюат количественно перенести в мерную колбу на 100 мл, довести до метки дистиллированной водой. Колонку после работы регенерировать (см. пункт)
9. Определение SO4 2- обратным комплексонометрическим титрованием [4]:
9.1. В колбу для титрования поместить 10 мл анализируемого раствора, 2-3 капли 2NHCl и прокипятить 2 – 3 минуты для удаления СО2 (если использовать прокипяченную дистиллированную воду, то кипячение смеси необязательно). Добавить 5 мл ВаCl2 (готовится растворением 0,5г ВаCl2 *2Н2 О и 0,1 г MgCl2 *6Н2 О в мерной колбе на 250 мл). Растворение проводить по методике, описанной в п. 1.3.
9.2. Оттитровывание избытка Ва2+ :
Раствор перемешать, охладить. Добавить 5 мл аммонийного буфера и немного сухого эриохрома. Смесь оттитровать стандартным раствором ЭДТА от вино-красной до синей окраски. Определить V1 титранта.
9.3 Холостой опыт: вместо анализируемого раствора в колбу для титрования поместить 10 мл дистиллированной воды. Повторить п. 4.1. и 4.2. Определить V2 титранта.
MgSO4 + ВаCl2 → ВаSO4 ↓ + MgCl2
Ва2+ изб. + H2 Ind → ВаInd + 2Н+
![]()
![]() HOOCH2
CCH2
COONa
HOOCH2
CCH2
COONa
![]()
![]() ВаInd + N─ CH2
─ CH2
─ N → H2
Ind . NaOOCH2
C CH2
COOH
ВаInd + N─ CH2
─ CH2
─ N → H2
Ind . NaOOCH2
C CH2
COOH
![]()
![]() -
OOCH2
C CH2
COONa
-
OOCH2
C CH2
COONa
+ N─ CH2 ─ CH2 ─ N
![]()
![]() NaOOCH2
C CH2
COO-
NaOOCH2
C CH2
COO-
Ва
Таблица 4
Определение сульфат-ионов
| № | Vопр | V1титранта | V2титранта | Условия титрования |
1 2 3 |
10 10 10 |
V1 V2 V3 V1титранта ср |
V1 V2 V3 V2титранта ср |
1. Обратное, индикаторное титрование. 2.Титрант – ЭДТА. 3.В колбе для титрования 10 мл элюата + 5 мл ВаCl2 + 5 мл аммонийного буфера + эриохром. 4. рН=10. 5. Титровать энергично перемешивая от винно-красной до синей окраски. Объем избыточной капли не вычитать. |
Расчеты:
1. СN (SO4 2- ) = CN (ЭДТА)*( V2 (ЭДТА) – V1 (ЭДТА) )/ Vопр
2. T(ЭДТА/SO4 2- ) = CN (ЭДТА)* Mэ (SO4 2- ) /1000
3. T(ЭДТА/ (MgSO4 )= CN (ЭДТА)* Mэ (MgSO4 )/1000
4. ω%пр (SO4 2- ) = [T(ЭДТА/SO4 2- )* V(ЭДТА) * Vразв * 100] /gпр . ( MgSO4 )*Vоп
5. ω%теор (SO4 2- ) = М (SO4 2- ) / М(MgSO4 ) = 96,062 /120,36=0,7981 или79,81 %
6. Dабс = ωпр (SO4 2- ) – ω теор (SO4 2- )
7. Dотн . = Dабс *100% / ωтеор (SO4 2- )
8. ω%пр (MgSO4 )= T(ЭДТА/ MgSO4 )*V(ЭДТА)* Vразв * 100] / gпр . ( MgSO4 )*Vоп
9. Dабс (MgSO4 ) = ωпр (MgSO4 )- ωтеор (MgSO4 )
10. Dотн . (MgSO4 ) = Dабс (MgSO4 )*100% / ωтеор (MgSO4 )
Глава 2. Химический эксперимент
2.1. Задачи эксперимента
· Освоить методику получения сульфата магния.
· Отработать практические навыки по неорганическому синтезу.
· Провести качественный и количественный анализ полученного соединения, тем самым закрепить теоретические и практические знания по аналитической химии.
· Отработать навыки самостоятельной работы в химической лаборатории.
2.2. Синтез сульфата магния
2.2.1. Методика синтеза
По методике, описанной в практикуме Корякина и Ангелова [1], MgSO4 можно получить, растворяя карбонат магния в серной кислоте:
MgCO3 + H2 SO4 → MgSO4 + CO2 ↑+ H2 O
Вгорячую30%-ную серную кислоту вносят при энергичном перемешивании MgCO3 (технический) до прекращения вспенивания жидкости. Отфильтрованная проба жидкости не должна давать красного окрашивания с NH4 SCN, в противном случае прибавляют еще MgCO3 . Раствор фильтруют и фильтрат оставляют кристаллизоваться на холоду. На следующий день, выпавшие кристаллы отсасывают на воронке Бюхнера, промывают небольшими количеством ледяной воды и перекристаллизовывают из воды (на 100 г соли берут 40 мл воды).
2.2.2. Расчет синтеза
Таблица 5
Характеристика исходных веществ
| Вещество | Mr | В кристаллическом виде | ||
t плавления 0 С |
растворимость, г/100 г Н2 О | |||
| При 200 С | при 1000 С | |||
| MgCO3 | 84,32 | >350 | Сл.р | - |
| MgSO4 | 120,36 | 1127 | 33,7 | 50 |
| MgSO4 *H2 O | 138,383 | 200 разл | - | 68,3 |
| MgSO4 *6H2 O | 22,458 | - | 44,5 | 73,4 |
| MgSO4 *7H2 O | 246,475 | 50 | 35,5 | - |
H2 SO4 |
98,07 | 10,37 | ∞ | ∞ |
Полностью нам нужно получить 10 г чистого вещества, а практический выход составляет 67%, то, исходя из формулы ω = mпр /mтеор , выразим
mтеор = mпр / ω
Сделаем расчет:
mтеор =10 г/0,67 = 14,97 г = 15 г
Подставим полученное число в уравнение реакции и по пропорции высчитаем массы реагирующих веществ:
ху 15 г
MgCO3 + H2 SO4 → MgSO4 + CO2 +H2 O
М 84,314г/моль 98,07г/моль 120,36г/моль
х = m(MgCO3 ) = 84,314 * 15 / 120,36 = 10,50 (г)
у = m(H2 SO4 ) = 98,07 * 15 / 120,36 = 12,22 (г)
Для синтеза нужно взять 10,50 г карбоната магния и 12,22 г серной кислоты. Но т.к. H2 SO4 – это жидкость, то нужно пересчитать массу на объем:
ω = mв-ва / mр-ра → mр-ра = mв-ва / ω = 10,5/0,3 = 40,7 (г)
ρ = mр-ра / Vр-ра → Vр-ра = mр-ра / ρ = 40,7/1,118 = 36,4 (мл)
Для проведения эксперимента будем использовать эти рассчитанные количества исходных веществ.
2.2.3. Подготовка к синтезу
Необходимое оборудование и реактивы:
Приготовление 50 мл раствора 30%-ной серной кислоты:
- По справочнику [2] определяем плотность 30%-ной серной кислоты:
ρ = 1,118 г/мл
mр-ра =50*1,118 = 55,9 (г)
mвещ-ва =0,3*55,9=16,77 (г)
- Ареометром измеряем плотность концентрированной кислоты, имеющейся в лаборатории:
ρ = 1,830 г/мл
- Находим по справочнику [2] ωконц. = 93,64 %
- Рассчитываем необходимый объем концентрированной кислоты:
mр-ра = 16,77/ 0,9364 = 17,91(г)
Vр-ра = 17,91/ 1,830 = 9,8(мл)
Для проведения синтеза нужно 9,8 мл концентрированной серной кислоты и довести до 50 мл водой, соблюдая правила техники безопасности.
2.2.4. Техника безопасности при работе с серной кислотой
Если необходимо развести концентрированную серную кислоту, то нужно наливать ее тонкой струйкой в воду и непрерывно перемешать. Если сделать наоборот, то кислота, в которую попала вода, моментально вскипит, и произойдет взрыв пара с выбросом капелек концентрированной кислоты, которые могут попасть на руки и лицо, что вызовет мгновенное их обугливание кислотой.
Другое правило – не работать с серной кислотой в хорошей одежде. Как бы хорошо ни укутываться в лабораторный халат или любую другую одежду, кислота все равно испортит все.
Третье правило – правило организации лабораторий. Серную кислоту нельзя сливать в канализацию. Если вы это делать постоянно, канализация превратится в дырявое решето. Слабоконцентрированная серная кислота хорошо уничтожает чугунные и пластиковые канализационные трубы. Если все таки сливать кислоту в канализацию, то это надо делать следующим образом. Пустить максимальный поток холодной воды и потихоньку слить кислоту, после чего держать поток воды еще минут 5-10, чтобы смыть ее остатки.
При попадании кислоты внутрь, ее необходимо как можно быстрее оттуда удалить, самый простой способ - вызвать рвотный рефлекс, вставив два пальца в рот и нажать на язык как можно дальше в горле, если это не получается, быстро выпить как можно больше воды и попытаться еще раз. После того как кислота удалена, надо выпить как можно больше воды и повторить рвотный рефлекс, желательно не один раз, чтобы смыть остатки кислоты с желудка и пищевода. После всех этих процедур необходимо срочно показаться врачу.
При попадании 10% и более концентрированной кислоты на кожу, кислоту надо быстро смыть под струей холодной воды, причем именно струей, потом обмыть теплой водой и промокнуть туалетной бумагой. Вафельное или махровое полотенце очень грубый материал и при вытирании ими поврежденной кожи, истонченная кислотой кожа может порваться или сняться вообще. Если кислота успела разъесть кожу и появилась обширная рана, рану необходимо закрыть стерильной медицинской салфеткой пропитанной перекисью. Очень важно, для того чтобы потом было меньше рубцов не дать ране высохнуть или не дать прилипнуть салфетке. Для того, чтобы салфетка дольше не высохла, ее можно накрыть пергаментом или калькой (не бумагой, бумага ее высушит быстрее), после чего рану забинтовать и срочно отправиться к врачу, чтобы он обработал рану специальными средствами (от гнойников, грибков и активизирующих заживление). Если рана небольшая, ее необходимо намазать антисептиком или заживляющим составом, не в коем случае не следует мазать поврежденные места йодом или зеленкой, а так же использовать для промывки раны спирт и спиртосодержащие препараты. Спирт вызовет ожог, с последующим осложнением заживления и может вызвать болевой шок у пострадавшего.
При работе с оборудованием При работе с техническими весами: убедившись в исправности розетки, чистоте поверхности, добиться равновесия и взвешивать вещества в стакане или в бумажной капсуле. По окончании операции взвешивания нужно выключить весы, а затем питание.При работе с электрической плиткой: перед началом работы с плиткой необходимо проверить исправность розетки, вилки и цельность изоляции провода. Не допускать соприкосновения провода с греющей поверхностью плитки. После окончания работы обязательно выключить плитку. Не выключенный прибор может привести к пожару.2.2.5. Синтез вещества
Взвесить на технических весах 10,5 г карбоната магния, растворить в 36,4 мл 30% серной кислоты. Происходит бурное вспенивание, поэтому необходимо добавлять карбонат магния маленькими порциями при постоянном перемешивании и под вытяжным шкафом. Раствор отфильтровать через складчатый фильтр. При фильтровании необходимо дополнительно немного нагревать раствор, для того чтобы растворить образовавшиеся на стенках воронки и фильтра кристаллов, чтобы избежать больших потерь вещества. Отфильтрованная проба жидкости не дала красного окрашивания с NH4 SCN, что свидетельствует об отсутствии катионов железа. Раствор фильтруем и оставляем кристаллизоваться в шкафу. Слить маточный раствор с выпавших кристаллов в отдельную пробирку. Полученные кристаллы поместить между листами фильтровальной бумаги и высушить. После высушивания полученные кристаллы пересыпали в склянку для реактивов №1, которую и будем в последующем использовать для проведения дальнейшего анализа. Мы получили шести-водный сульфат магния MgSO4 *6H2 O. Оставшийся маточный раствор (V= 25 мл), который в связи с хорошей растворимостью сульфата магния при комнатной температуре (см. таблицу свойств) содержит его в растворенном состоянии, упариваем на водяной бане до появления пленки на поверхности жидкости и оставляем кристаллизоваться в шкафу. Полученные кристаллы просушить между листами фильтровальной бумаги, затем высушить в сушильном шкафу при температуре 120°С. Полученные мелкие кристаллы поместить в ступку и растереть, после чего взвесить и пересыпать в банку для реактивов №2. Так как мы сушили при 120°С, то получили смесь гидратов с 1-ой, возможно 2-мя молекулами воды.
Рассчитаем, какой получился выход вещества в банке для реактивов №1:
ω% (вещества) = mпр. * 100% / mтеор. Так как мы должны были получить безводный сульфат магния MgSO4 (M = 120,36 г/моль), а получили шестиводный MgSO4 *6H2 O (M = 228,458 г/моль), то необходимо сделать пересчет:mтеор = 15 *228,458/120,36 = 28,5 гω% (вещества) = 5,3 * 100% / 28,5 = 18,6%Практический выход в банке для реактивов №2:Так как мы должны были получить безводный сульфат магния MgSO4 (M = 120,36 г/моль), а получили смесь гидратов, то необходимо сделать пересчет:mтеор = 15 * 228,458/120,36 = 28,5 гω% (вещества) = 11,4 * 100% / 28,5 = 40% Практическая масса получилась много меньше, чем теоретическая, что связано с потерей вещества в результате фильтрования, так как в процессе фильтрования сульфат магния начал кристаллизоваться на стенках воронки и фильтра; так же в связи с его хорошей растворимостью в воде даже при комнатной температуре и большая часть вещества растворилась и перешла в в маточный раствор; много потерь было и при просушивании кристаллов между листами фильтровальной бумаги.Анализ синтезированного соединения и возможных примесей
Подготовка к анализу:
1. оборудование и реактивы:
Оборудование:
· штатив с пробирками
· спиртовка
· держатель
· шпатель
· предметное стекло
· микроскоп
· стеклянная палочка
· универсальная индикаторная бумага
Реактивы (готовили лаборанты):
1. Гидроксид натрия –NaOH (2N);
2. гексацианоферрат (III) калия (красная кровяная соль) - К3 [Fe(CN)6 ];
3. серная кислота - H2 SO4 (2N);
4. карбонат калия - К2 CO3 ;
5. азотная кислота - HNO3 (2N);
6. перманганат калия – KMnO4 ;
7. уксусная кислота - CH3 COOН (2N);
8. хлорид кальция – CaCl2 ;
9. медь- Cu;
10. этиловый спирт C2 H5 OH;
11. сульфат аммония (NH4 )2 SO4 ;
12. ацетат аммония CH3 COONH4 ;
13. иодид калия KI;
14. сульфат калия K2 SO4 ;
15. персульфат аммония NH4 S2 O8 ;
16. нитрат серебра AgNO3 ;
17. пероксид водорода H2 O2 ;
18. гидрофосфат натрия Na2 HPO4 ;
19. карбонат калия К2 СО3 ;
20. дитизон C13 H12 N4 S;
21. карбонат натрия Na2 CO3 .
Оборудование:
· Весы технические
· Весы аналитические
· Колбы мерные на 50 и 100 мл
· Колба для титрования на 50 мл
· Стеклянная воронка
· Пробка резиновая для мерных колб
· Стеклянная палочка
· Ионообменные колонки с катионитом КУ-2 в Н – форме;
· Пипетки на 2, 5, 10 мл
· Пипетка глазная
· Шпатель
· Бутылки для хранения реактивов
· Универсальная индикаторная бумага
Реактивы:
1. Соляная кислота HCl конц (если есть, то 2 N);
2. Эриохром;
3. Сульфат магния MgSO4 *7H2 Oкр ( если есть, то из фиксанал на сульфат магния);
4. ЭДТА кр (или из фиксанала);
5. Раствор аммиака NH3*H2 O 25% и хлорид аммония NH4 Cl кр (если есть, то аммонийный буфер);
6. Эриохром;
7. Хлорид бария BaCl2 кр;
8. хлорид магния MgCl2 кр.
Все реактивы и растворы были приготовлены лаборантами.
2. Приготовление и стандартизация растворов для комплексонометрического определения Mg2+ и SO4 2- .
2.1. Приготовление анализируемого раствора:
Приготовить 50 мл 0,1N раствора MgSO4
1) Расчет навески:
1. fэ =1/2
2. Мэ = 120,36 * ½ = 60,18 г/моль
3. gт =CN *Vл *Mэ =0,1моль/л*0,050л*60,18г/моль=0,3009 г
2). Взятие навески (сначала на ТВ, затем на АВ):
mпуст.кап =0,3054 г
mкап.с в-вом теор. = 0,3054 + 0,3009= 0,6063 (г)
mкап.с в-вом = 0,6162 г
mкап.с ост. =0,3060 г
gпракт. =0,3102 г
3). Расчет практической концентрации:
CN = gпракт / Vл *Mэ = 0,3102 г/0,05 л*60,18 г/моль=0,1030 моль/л
Приготовить 50 мл 0.1 Nраствора MgSO4 *7H2 O.
Готовили из фиксанала ( 1 литр раствора):
1. Заполнить мерную колбу (V = 1 л) на 1/3 объема дистиллированной водой.
2. разбить капсулу фиксанала с навеской и высыпать ее в мерную колбу через сухую воронку. Смыть большим количеством воды остатки навески из капсулы и с воронки и горлышка колбы.
3. Растворить соль. Если вещество растворяется плохо, колбу слегка нагреть на плитке до полного растворения вещества.
4. Охладить колбу до комнатной температуры. Довести объем раствора в колбе дистиллированной водой до метки по нижней границе мениска (последнюю каплю воды добавлять глазной пипеткой).
5. Закрыть колбу пробкой и перемешать 15-20 раз. Содержимое колбы вылить в чистую посуду.
2.3. Приготовление рабочего раствора методов прямого и обратного комплексонометрического титрований:
Приготовить 50 мл 0,1 Nраствора ЭДТА.
Готовили из фиксанала 1 литр раствора( метод описан в пункте 2.2.).
2.4. Приготовление аммонийного буфера: готовили лаборанты.
2.5. Приготовление раствора хлорида бария ( BaCl 2 ): готовили лаборанты.
2.6. Приготовление 50 мл 4 N HCl : готовили лаборанты.
Стандартизация рабочего раствора
| № | Vуст.вещ-ва СN = 0,1 |
VЭДТА | Условия титрования |
1 2 3 |
2 2 2 |
2 2 2 Vср = 2 |
1. Прямое, индикаторное титрование. 2.Титрант – ЭДТА. 3.В колбе для титрования 2 мл MgSO4 ∙7H2 O+ 2мл аммонийного буфера + эриохром. 4. рН=10. 5. Титровать энергично перемешивая от винно-красной до синей окраски. Объем избыточной капли не вычитать. |
Расчеты:
1. CN (ЭДТА) = CN (MgSO4 ∙7H2 O)*V(MgSO4 ∙7H2 O)/V(ЭДТА) = 0,1моль/л * 2 мл / 2 мл = 0,1 (моль/л);
2. Т(ЭДТА) = CN (ЭДТА)*Мэ(ЭДТА)/1000 = 0,1моль/л * 186,12 г/моль / 1000 = 0,01861 (г/мл);
3. Т(ЭДТА/MgSO4 ∙7H2 O) = CN (ЭДТА)*Мэ(MgSO4 ∙7H2 O)/1000 = 0,1моль/л * 123,235 г/моль / 1000 = 0,01232 (г/мл).
Качественный анализ сульфата магния и возможных примесей:
Анализ проводится согласно схемы 1по методике, описанной на стр 13 - 27.
Таблица 7
Качественный анализ сульфата магния
| № | исследуемое вещество | реагент | аналитический эффект | вывод |
| 1 | 2 | 3 | 4 | 5 |
| 1 | Сухая соль | Растворение в воде | Бесцветная прозрачная жидкость | Возможно присутствие только растворимых соединений |
| 2 | Раствор 1 | Дробно: H2 SO4 +C2 H5 OH, | Бесцветная однородная жидкость |
Ca2+ - нет; Pb2+ - нет |
| 3 | Раствор 1 | K2 CO3 | Белый мутный осадок | Катионы тяжелых металлов |
| 1 | 2 | 3 | 4 | 5 |
| 4 | Раствор 5 | Дробно: Cu + H2 SO4 3 |
Бесцветная однородная жидкость |
NO3 - - нет 5 |
| 5 | Раствор 5 |
Дробно: CaCl2 , микрокрист |
Белый осадок в виде игл (под микроскопом) | SO4 2- - есть |
6 |
Раствор 5 |
Дробно: KMnO4 + H2 SO4 , йодокрахмальная бумажка |
Бесцветная однородная жидкость |
Сl- - нет |
| 7 | Осадок 9 |
HNO3 |
Осадок растворяется, бесцветная однородная жидкость | Тяжелые металлы перешли в раствор |
| 8 | Раствор 10 | H2 SO4 + C2 H5 OH, центрифугируем |
Бесцветная однородная жидкость | Ca2+ - нет; Pb2+ - нет; В растворе тяжелые металлы |
9 |
Раствор 11 | Дробно: K3 [Fe(CN)6 ] |
Бесцветная однородная жидкость | Fe2+ - нет |
| 10 | Раствор 11 | NaOH изб | Белый мутный осадок | Катионы тяжелых Ме |
| 1 | 2 | 3 | 4 | 5 |
| 11 | Осадок 5.1. | H2 O2 , t,центрифуг. | Белый осадок |
Катионы тяжелых металлов |
| 12 | Осадок 6 | NH4 Cl насыщ, центрифуг. |
Бесцветная однородная жидкость | |
| 13 | Раствор 8 | Na2 HPO4 + NH4 Cl | Осадок |
Mg2+ - есть |
По результатам качественного анализа обнаружены Mg2+ и SO4 2- . Примесей в полученном веществе не обнаружено, сульфат магния химически чист.
1. Количественное определение Mg2+ проводится по методике, описанной на стр. 33 – 34:
Метод прямой комплексонометрии:
Титрование анализируемого раствора. Таблица 8
| № | Vсоли | VЭДТА | Условия титрования |
1 2 3 |
2 2 2 |
1,1 1,1 1,1 1,1 |
1. Прямое, индикаторное титрование. 2.Титрант – ЭДТА. 3.В колбе для титрования 2 мл MgSO4 + 2 мл аммонийного буфера + сухой эриохром. 4. рН=10. 5. Титровать энергично перемешивая от винно-красной до синей окраски. Объем избыточной капли не вычитать. |
Расчеты:
1. CN (Mg2+ ) = CN (ЭДТА)*V(ЭДТА)/Vопр = 0,1*1,1 /2 = 0,0550 (моль/л);
Полученная концентрация не сходится с теоретической, что может говорить о том, что полученная соль кристаллогидрат или в соле есть примеси, которые ничтожно малы.
2. Т(ЭДТА/Mg2+ ) = CN (ЭДТА)*Мэ(Mg2+ )/1000 = 0,1*12,1525/1000 = 0,001215;
3. Т(ЭДТА/MgSO4 ) = CN (ЭДТА)*Мэ(MgSO4 )/1000 = 0,1*60,18/1000 = 0,006018;
4.ω%пр (Mg2+ ) = [T(ЭДТА/ Mg2+ )*V(ЭДТА) * Vразв * 100] / [gпр. ( MgSO4 )*Vоп ] = [0,001215*1,1*50*100]/[0,3066*2] = 10,77%;
5. ω%теор (Mg2+ ) = М(Mg2+ )/М(MgSO4 ) = 24,305/120,36=0,2019 или 20,19%;
6. Dабс = ωпр (Mg2+ ) - ωтеор (Mg2+ ) = 10,77% - 20,19% = 9,41%;
7. Dотн. = Dабс *100% / ωтеор. ( Mg2+ ) = 9,41*100/20,19 = 46,65%;
8. ω%пр (MgSO4 ) = [T(ЭДТА/ MgSO4 ) *V(ЭДТА) *Vразв *100]/[gпр. (MgSO4 )*Vоп ] = [0,006018*1,1*50*100]/[0,3102*2] = 53,35%;
9. Dабс = ωпр (MgSO4 ) - ωтеор (MgSO4 ) = 53,35% – 100% = -46,65%;
10. Dотн. = Dабс *100% / ωтеор. ( MgSO4 ) = 46,65*100/100 = 46,65%.
Исходя из полученных расчетов, можно предположить, что анализируемое вещество кристаллогидрат, так как разница между ω % (MgSO4 ) теор и ω % (MgSO4 )практ существенна. Найдем процентное содержание воды в кристаллогидрате и выведем его формулу:
11. ω % расч (H2 O)= 100% - 53,35% = 46,65 %;
12. ω % теор
(H2
O) =![]() ;
;
46,65 = ![]() ;
;
1801,53*n= 5615,17 + 840,42*n;
961,11*n= 5615,17;
n=5,84~6.
Значит, получился гексагидрат MgSO4 ∙ 6H2 O.
13. ω %
(Mg2+
)теор
=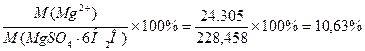 ;
;
14. Dабс = ω теор. ( Mg2+ )- ω пр. (Mg2+ )=10,63 – 10,77 = -0,14 %;
15. Dотн. = Dабс *100% / ω теор. ( Mg2+ )=0,14*100% /10,63 = 1,32 %.
| № | Vопр | VЭДТА | VЭДТА хол. | Условия титрования |
1 2 |
20 20 |
1,05 1,05 VЭДТАср 1,05 |
1,6 1,6 VЭДТАср 1,6 |
1. Обратное, индикаторное титрование.. 2. Титрант – ЭДТА. 3. В колбе для титрования 10 мл анализируемого раствора + 5 мл ВаCl2 + 5мл аммонийного буфера + эриохром. 4. рН=10. 5. Титровать энергично перемешивая от винно-красной до синей окраски. Объем избыточной капли не вычитать. |
2. Количественное определение SO4 2- .
Определение сульфат - ионов Таблица 9
Расчеты:
1. СN (SO4 2- ) = CN (ЭДТА)*( V2 (ЭДТА) – V1 (ЭДТАхолл.оп ) )/ Vопр = 0,1 моль/л*(1,6 мл – 1,05 мл)/10 мл = 0,003000 (моль/л);
2. CN (SO4 2- ) исх. р-ра = CN (SO4 2- )разб. р-ра *10 = 0,00300*10 = 0,03000 (моль/л);
3. T(ЭДТА/SO4 2- ) = CN (ЭДТА)*Mэ (SO4 2- ) /1000 = 0,1моль/л*48,031 г/моль/1000 = 0,004803 (г/мл);
4. T(ЭДТА/ (MgSO4 )= CN (ЭДТА)*Mэ (MgSO4 )/1000 = 0,1 моль/л*60,18 г/моль/1000 = 0,006018 (г/мл);
Прежде чем рассчитывать ω % ( SO4 2- )практ и ω % (MgSO4 ) практ нужно учесть, что для титрования использовалась не вся навеска. Рассчитаем её. Сначала навеску g= 0,3102 г растворили в мерной колбе на 50 мл, затем 10 мл пропустили через колонку, т.е. навеску, используемую для анализа можно рассчитать по формуле:
5. g' = g *Vопр /Vколбы = 0,3102*10 мл/50 мл = 0,06204 (г);
6. ω% (SO4
2-
)практ
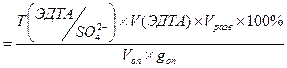 = 0,004803*0,55*100*100/(10*0,06204) = 42,57 %;
= 0,004803*0,55*100*100/(10*0,06204) = 42,57 %;
7. ω%(MgSO4
)практ
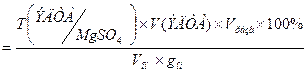 =
=
0,006018*0,55*100*100/(10*0,06204) = 53,35 %;
Исходя из полученных расчетов, можно сделать вывод, что анализируемое вещество кристаллогидрат, так как разница между ω% (MgSO4 )теор и ω% (MgSO4 )практ существенна.
8. ω % расч (H2 O)= 100% - 53,35% = 46,65 %;
9. ω % расч
(H2
O) =![]() ;
;
46,65=![]() ;
;
1801,53*n= 5617,17 + 840,42*n;
961,11*n = 5615,7;
n= 5,84~6.
Значит, получился гептагидрат MgSO4 ∙ 6H2 O.
10. ω %( SO4
2-
) теор
=![]() ;
;
11. Dабс = ω теор. ( SO4 2- )- ωпр . ( SO4 2- )= 42,10- 42,57 = -0,47%;
12. Dотн . = Dабс *100% / ω теор. ( SO4 2- )=0,47*100 /42,10 = 1,12 ~ 1,1 %.
Можно рассчитать, сколько практически содержится воды:
13. ω%расч (H2 O) = 100% - ω% ( SO4 2- )практ - ω%( Mg2+ )практ = 100 – 42,57 – 10,77 = 46,66 %;
14. ω%расч (MgSO4 ) = (53,35+53,35)/2 = 53,35%;
15. ω%теор (MgSO4 ∙ 6H2 O) = 53,35 + 46,66 = 100,01 %;
16. Dабс = ω теор. ( MgSO4 ∙ 6H2 O)- ω пр. ( MgSO4 ∙ 6H2 O)=100 – 100,01 = 0,01%;
17. Dотн. = Dабс *100% / ω теор. ( Mg2+ ) = 0,01*100% /100 = 0,01%.
Итоговая таблица:
Таблица 10
W%. теор (MgSO4 ∙ 6H2 O) |
W%.пр. | Dабс. % | Dотн. % | |
| Mg2+ | 10,63 | 10,77 | 0,14 | 1,32 |
| SO4 2- | 38,70 | 38,97 | 0,27 | 0,7 |
| MgSO4 ∙ 6H2 O | 100 | 100,01 | 0,01 | 0,01 |
Полученные в ходе эксперимента результаты отличаются от теоретически расчитанных. Следовательно, выбранные для анализа методы дают хорошие результаты. Но все равно имеется погрешность. Погрешности эксперимента могут иметь как систематический (несовпадение КТТ и ТЭ), так и случайный характер (связанны с тем, что не проводилась калибровка посуды, а также погрешности измерения объема и взвешивания). Вероятно, источниками погрешностей могут быть: погрешности метода, или инструментальные погрешности, погрешности образцов сравнения, реактивная погрешность, т.к. все применяемые реактивы не могут быть абсолютно чистыми.
Заключение
В ходе данной работы по литературным источникам была выбрана методика синтеза соединения, составлена методика его качественного и количественного определения, а также возможных примесей. Был проведен синтез этого соединения, его качественное и количественное определение. По результатам качественного анализа не было обнаружено примесей, следовательно синтез был проведен чисто. По результатам количественного анализа получено:
ω%пр (Mg2+ )= (10,77 ± 0,14)
ω%пр. (SO4 2- )= (38,97 ± 0,27)
ω%пр (MgSO4 *6H2 O)= (100,01 ± 0,01)
Погрешности могут быть как случайными, так и систематическими.
Методы, применяемые в данной работе не требуют специальной аппаратуры, поэтому эти методы достаточно распространены, и часто используются в химической практике. Простота их применения и достаточная точность делают их оптимальными для наших условий Достаточная точность достигается аккуратностью выполнения. На сегодняшний день существует огромное количество различных методов и методик проведения анализа, имеющих очень небольшую погрешность, но они достаточно сложны в применении, и требуют специальных знаний, а также в большинстве случаев дорогой аппаратуры.
Библиографический список
1. Корякин Ю.В., Ангелов И.И. Чистые химические вещества. 4-е изд., перераб. – М.: Химия, 1974. – 1575 с.
2. Лурье Ю.Ю. Справочник по аналитической химии. – М.: Химия, 1983. – 200 с.
3. Попова Л.Ф. Качественный анализ. Лабораторный практикум по аналитической химии: Методическая разработка. 2-е изд., испр. – Архангельск: Изд-во ПГУ, 2005 – 146 с.
4. Попова Л.Ф. Количественный анализ. Лабораторный практикум по аналитической химии: Методическая разработка. 2-е изд., испр. – Архангельск: Изд-во ПГУ, 2005 – 60 с.
5. Попова Л.Ф. Физико-химические методы анализа. Лабораторный практикум по аналитической химии: Методическая разработка. 2-е изд., испр. – Архангельск: Изд-во ПГУ, 2005 – 105 с.
6.Ключников И.Р. Неорганический синтез. – М.: Просвещение, 1971. –320 с. с ил.
7. Справочник химика. Основные свойства неорганических и органических соединений. 2-е изд., перераб. и доп. – М.: Химия, 1964. – Т.2. – 1168 с.
8. Химические свойства неорганических веществ: Учеб. пособие для вузов. 3-е изд., испр./ Р.А. Лидин, В.А. Молочко, Л.Л. Андреева; Под ред. В.А. Лидина. – М.:Химия,2000. 480 с.: ил.
9. Химическая энциклопедия в пяти томах. /Под ред. И.Л.Кнунянц – М.: издательство «Советская энциклопедия »,1988, - Т.1. – 1250 с.
10. http://www.alhim.com.ua/catalog/1819/good-82.html
11. http://www.bhz.kosnet.ru/Rus/Prod/Tech/Mg_sulf.html
12. http://www.chempack.ru/chemistry/details/magnesium_sulfate.html
Приложение 1
![]()
Сульфат магния
MgSO4 *6H2 O№1
Приложение 2
![]()
Сульфат магния
MgSO4 *6H2 O№2