| Скачать .docx |
Реферат: Легко- и трудногидролизуемые полисахариды
Реферат
Легко- и трудногидролизуемые полисахариды
Тамбов, 2009
Содержание
Введение
1. Методы определения редуцирующих веществ в гидролизатах
2. Определение легкогидролизуемых полисахаридов
3. Определение трудногидролизуемых полисахаридов
4. Определение массовой доли PB в гидролизатах по методу Макэна и Шоорля
5. Определение массовой доли PB в гидролизатах эбулиостатическим методом
6. Анализ гидролизатов методом газожидкостной хроматографии
Введение
Определение легко - и трудногидролизуемых полисахаридов в древесине основано на реакциях их гидролиза с последующим нахождением общего количества образовавшихся моносахаридов по редуцирующей способности. Для определения содержания отдельных моносахаридов в гидролизатах - гексоз и пентоз - используют хроматографический анализ.
Легко- и трудногидролизуемые полисахариды разделяют, используя различные условия гидролиза. Для гидролиза легкогидролизуемых полисахаридов применяют обработку древесины разбавленными минеральными кислотами, а для гидролиза трудногидролизуемых полисахаридов - обработку концентрированной кислотой при температуре 20...25°С. Для полного гидролиза полисахаридов древесины вместо серной кислоты можно использовать трифторуксусную кислоту.
Гидролиз полисахаридов разбавленной кислотой идет в гетерогенной среде, а гидролиз концентрированной кислотой - в гомогенной. В концентрированной кислоте полисахариды сначала набухают и растворяются, а затем уже происходит их гидролитическая деструкция. Однако в связи с недостатком воды в реакционной смеси продуктами гидролиза в концентрированной кислоте являются не моносахариды, а олигосахариды.
Они образуются, во-первых, при частичном гидролизе полисахаридов, а во-вторых, в результате реверсии моносахаридов - реакции обратной гидролизу. Для доведения гидролиза до конечной стадии - получения моносахаридов - проводят дополнительный гидролиз - инверсию. Раствор олигосахаридов в концентрированной кислоте разбавляют и кипятят в течение 3...5 ч.
При анализе малоизученного сырья рекомендуется проводить предварительные опыты для установления необходимой продолжительности стадии дополнительного гидролиза до достижения максимального выхода моносахаридов.
При слишком длительной стадии дополнительного гидролиза начинается распад моносахаридов, а также происходит реакция реверсии.
1. Методы определения редуцирующих веществ в гидролизатах
Для определения общего количества легко - или трудногидролизуемых полисахаридов находят концентрацию редуцирующих веществ в гидролизатах. Определение легко - и трудногидролизуемых полисахаридов по концентрации редуцирующих веществ не является вполне точным. В гидролизатах редуцирующими веществами будут не только моносахариды, но также и продукты их распада в кислой среде. В гидролизатах трудногидролизуемых полисахаридов PB состоят главным образом из глюкозы и небольших количеств маннозы, ксилозы, фруктозы. В гидролизатах легкогидролизуемых полисахаридов состав PB более разнообразен: глюкоза, манноза, галактоза, ксилоза, арабиноза, рамноза, глюкуроновая и галактуроновая кислоты, продукты распада моносахаридов. Все вещества имеют различную редуцирующую способность. Учесть эти различия при расчете результатов анализа практически невозможно. Поэтому принято концентрацию PB в гидролизатах определять чаще всего в пересчете на глюкозу. Для определения общего выхода PB пользуются методом Бертрана или эбулиостатическим методом, основанными на реакции окисления Сахаров медно-щелочным раствором, в результате которой двухвалентная медь Cu2+ переходит в одновалентную Cu+ и выпадает в осадок в виде оксида меди Cu2 O. В качестве медно-щелочного раствора используют реактив Фелинга, который получают непосредственно при проведении анализа смешиванием растворов сульфата меди и щелочного раствора сегнетовой соли, в результате чего получается растворимый комплекс, содержащий Cu2+ ,
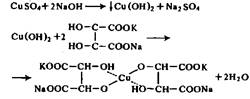
Упрощенно окисление альдоз реактивом Фелинга можно представить схемой
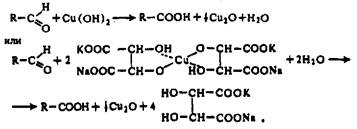
В действительности же окисление редуцирующих Сахаров медно-щелочным раствором представляет не одну определенную реакцию, а множество реакций, происходящих одновременно. При этом носителями редуцирующих свойств являются не только сами сахара, но и продукты их дальнейших превращений и распада. Таким образом, здесь нельзя ожидать стехиометрически протекающей реакции и написать точное уравнение реакции фактически невозможно. Поэтому для пересчета массы восстановленной меди в массу Сахаров пользуются эмпирическими таблицами.
Первоначально при определении PB методом Бертрана осадок Cu2 O взвешивали. Впоследствии количество восстановленной меди стали определять титриметрическими методами - прямого или обратного титрования.
В прямом методе осадок Cu2 O растворяют в растворе додекагидрата сульфата железа - аммония NH4 Fe2 · 12Н2 0 в присутствии серной кислоты. При этом Cu+ окисляется в Cu2+ , aFe3+ восстанавливается в Fe2+ по уравнению
![]()
Раствор FeSO4 титруют раствором перманганата калия
![]()
По количеству перманганата калия, израсходованного на реакцию окисления железа, вычисляют массу меди в Cu2 O и по эмпирической таблице находят массу Сахаров в пересчете на глюкозу.
Ввиду того, что оксид меди легко окисляется на воздухе, определение Сахаров приводит к неточным результатам. Кроме того, для метода Бертрана характерно большое число операций. При нагревании медно-щелочного раствора возможно его самовосстановление. Самовосстановление меди можно учесть, если провести контрольное определение.
Более точный и простой метод обратного титрования был разработан Макэном и Шоорлем. Этот метод основан на йодометрическом определении избытка реактива Фелинга.
![]() "
"
Самовосстановление в этом методе учитывают, так как расход реактива Фелинга определяют по разности между результатами контрольного и рабочего титрований.
В обоих методах взаимодействие PB с реактивом Фелинга происходит при свободном доступе кислорода воздуха, и необходимо пользоваться эмпирическими таблицами. Этих недостатков лишен эбулиостатический метод, основанный на прямом титровании горячего медно-щелочного раствора исследуемым раствором сахара в специальном приборе - эбулиостате, позволяющем производить анализ в токе водяного пара, т.е. без доступа воздуха. Эмпирические таблицы не требуются. Устанавливают титр медно-щелочного раствора по раствору глюкозы известной концентрации.
Общий выход PB при количественном гидролизе определяют как сумму редуцирующих веществ, полученных при определении легко - и трудногидролизуемых полисахаридов. Наиболее точное представление о качественном и количественном составе редуцирующих веществ гидролизатов получают с помощью хроматографических методов анализа.
2. Определение легкогидролизуемых полисахаридов
Методика анализа. Навеску воздушно-сухих опилок массой около 5 г помещают в коническую колбу вместимостью 500 см3 , добавляют 200 см3 2% -ной HCI и кипятят с обратным холодильником на электрической плитке в течение 3 ч. Для регулирования кипения под колбу подкладывают асбестовую сетку или колбу приподнимают над плиткой. Все опилки должны находиться в кислоте. Для этого после разогрева содержимое колбы осторожно перемешивают. По окончании гидролиза отфильтровывают опилки на воронку Бюхнера с бумажным фильтром с отсосом. Остаток на фильтре промывают горячей водой до отрицательной реакции на кислоту по индикатору метиловому оранжевому и используют для определения трудногидролизуемых полисахаридов. Фильтрат и промывные воды переносят в мерную колбу вместимостью 500 см3 , доводят раствор после охлаждения дистиллированной водой до метки и тщательно перемешивают. В полученном растворе определяют массовую долю PB в процентах.
Массовую долю легкогидролизуемых полисахаридов, % к абсолютно сухой древесине, рассчитывают по формуле
![]()
где с" - массовая доля PB в гидролизате легкогидролизуемых полисахаридов, %; V - объем гидролизата, V=500 см3 ; k" - коэффициент пересчета моносахаридов в полисахариды; g - масса абсолютно сухой навески древесины, г.
Коэффициент пересчета k" вычисляется на основании реакций гидролиза полисахаридов. Для пентозанов 1гл -132/150=0,88, а для гексозанов A1 = 162/180 = 0,90, где 132 и 162 - молекулярные массы звеньев соответствующих полисахаридов, а 150 и 180 - молекулярные массы пентоз и гексоз. Принимая содержание гексоз и пентоз в гидролизатах древесины хвойных пород примерно равным, для расчета используют значение коэффициента £, = 0,89; для гидролизатов древесины лиственных пород берут = 0,88.
3. Определение трудногидролизуемых полисахаридов
Методика анализа. Остаток древесины после гидролиза легкогидролизуемых полисахаридов и промывки количественно переносят с фильтра в стакан вместимостью 100 см3 и подсушивают при 50...60°С примерно до воздушно-сухого состояния, а затем обрабатывают 35...40 см3 80% -ной H2 SO4 при комнатной температуре в течение 3 ч, периодически перемешивая стеклянной палочкой. Смесь из стакана количественно переносят в коническую колбу вместимостью 1000 см3 , смывая дистиллированной водой в количестве 600 см3 . Колбу присоединяют к обратному холодильнику и кипятят на электрической плитке в течение 3 ч. После окончания дополнительного гидролиза фильтруют раствор через воронку Бюхнера с бумажным фильтром с отсосом. Остаток на фильтре промывают небольшими порциями горячей воды до отрицательной реакции на кислоту по индикатору метиловому оранжевому. Фильтрат и промывные воды переносят в мерную колбу вместимостью 1000 см3 . После охлаждения доводят объем раствора до метки дистиллированной водой и тщательно перемешивают.
Из полученного раствора отбирают пипеткой 50 см3 в мерную колбу на 100 см3 и осторожно при постоянном перемешивании нейтрализуют 20% -ным раствором NaOH по метиловому оранжевому. Объем раствора доводят до метки дистиллированной водой. В нейтрализованном растворе определяют концентрацию PB.
Массовую долю трудногидролизуемых полисахаридов, % к абсолютно сухой древесине, рассчитывают по формуле
![]() "
"
где ст - массовая доля PB в разбавленном нейтрализованном гидролизате, %; V - общий объем кислого гидролизата, V= = 1000 см3 ; з - разбавление гидролизата при нейтрализации, л= 100/50 = 2; kT - коэффициент пересчета моносахаридов в полисахариды, равный 0,90; g - масса абсолютно сухой навески древесины, г.
Коэффициент пересчета берут равным 0,90, поскольку основным сахаром в гидролизате трудногидролизуемых полисахаридов является глюкоза.
4. Определение массовой доли PB в гидролизатах по методу Макэна и Шоорля
Для получения реактива Фелинга готовят два раствора А - 69,3 г CuSO4 5Н2 0 в 1 дм3 водного раствора; Б - 346 г сегнетовой соли и 100 г NaOHв 1 дм3 водного раствора.
Методика анализа. В коническую колбу вместимостью 250 см3 вливают пипеткой 10 см3 раствора А, затем 10 см3 раствора Б и 20 см3 гидролизата легкогидролизуемых полисахаридов или нейтрализованного гидролизата трудногидролизуемых полисахаридов. Смесь разбавляют дистиллированной водой до общего объема 50 см3 и хорошо перемешивают. Ставят колбу на горячую включенную электроплитку, нагревают смесь до кипения в течение 3 мин и кипятят точно 2 мин, считая с момента появления первого пузырька на поверхности раствора.
Под колбу рекомендуется подложить асбестовую пластинку с круглым вырезом диаметром около 6 см. Кипение должно быть умеренным, чтобы объем жидкости в колбе оставался примерно постоянным. Для уменьшения испарения в горло колбы вставляют маленькую конусообразную стеклянную воронку.
При недостатке реактива Фелинга, о чем свидетельствует исчезновение синей окраски раствора после кипячения, объем пробы гидролизата уменьшают, добавив при разбавлении соответствующий объем воды. По окончании кипячения колбу быстро охлаждают холодной водой до 25°С, добавляют раствор KIи 10 см3 25% -ной H2 SO4 и сразу же при непрерывном перемешивании титруют выделившийся иод раствором тиосульфата натрия концентрацией 0,1 моль/дм3 до перехода коричневой окраски в светло-желтую.
Затем добавляют 10 см3 0.5...1% -ного раствора крахмала и медленно дотитровывают раствор до полного исчезновения синей окраски.
Раствор остается окрашенным в кремовый цвет вследствие образования иодида меди. В аналогичных условиях, но без добавления раствора сахара, проводят контрольный опыт. По разности расходов раствора Na2 S2 O3 в контрольном и рабочем опытах, а, см3 , с помощью эмпирической таблицы находят количество сахара в пробе гидролизата, взятой на анализ, Ь, мг.
При анализе трудногидролизуемых полисахаридов расчет ведут на глюкозу, а при анализе гидролизата легкогидролизуемых полисахаридов - на ксилозу и маннозу. Затем рассчитывают массовую долю PB в гидролизате сл или ст ,%, по формуле
![]()
где b - количество сахара в пробе гидролизата объемом у, см1 , найденное по таблице, мг.
5. Определение массовой доли PB в гидролизатах эбулиостатическим методом
Проводят титрование медно-щелочного раствора с известным титром исследуемым раствором сахара без доступа воздуха в токе водяного пара. Титр медно-щелочного раствора устанавливают по глюкозе. Прибор для титрования состоит из внешнего сосуда - конической колбы / и внутреннего сосуда - эбулиостата 1 с впаянной в его боковую стенку трубкой, доходящей почти до дна сосуда. Суженный конец эбулиостата вставлен в колбу / на резиновой пробке и имеет отверстие для выхода пара 3. В резиновую пробку вставлена стеклянная трубка 4 для отвода избытка пара из колбы /. Струю пара регулируют винтовым зажимом на резиновой трубке, надетой на конец стеклянной трубки. Во время титрования верхний конец эбулиостата закрывают резиновой пробкой, в которую вставлен конец бюретки 5 для раствора сахара. Реагирующая жидкость в эбулиостате обогревается паровой рубашкой и пропусканием пара через жидкость.
Индикатором при титровании служит метиленовый голубой, который в окислительной среде имеет синий цвет, а в восстановительной бесцветен. Для предотвращения выпадения осадка оксида меди в медно-щелочной раствор добавляют желтую кровяную соль калия, которая образует с Cu2 O растворимое комплексное соединение.
Для проведения анализа приготовляют два раствора: А - 10 г CuSO4 ·5З2 0 и 0,04 г метиленового голубого растворяют в воде в мерной колбе на 1 дм3 доведением до метки; Б - 50 г сегнетовой соли растворяют в воде, отдельно растворяют в воде 4 г желтой кровяной соли, переносят оба раствора в мерную колбу на 1 дм \ добавляют 75 г NaOH в виде 50% -ного водного раствора и доводят водой до метки.
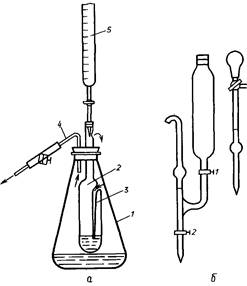
Прибор для определения PB эбулиостатическим методом и дозаторы растворов. Дозатор для отмеривания раствора А представляет собой два сообщающихся сосуда. Сосуд вместимостью около 100 см3 служит для хранения раствора А и сосуд-пипетка для его отмеривания. На верхнем и нижнем отводах сосуда-пипетки нанесены метки, соответствующие объему 5 cmj . Для отмеривания раствора А открывают кран / и заполняют сосуд-пипетку до верхней метки. Затем кран закрывают. При отмеривании раствора А и заливании его в эбулиостат открывают кран 2 и спускают раствор до нижней метки. Второй дозатор для отмеривания раствора Б представляет собой пипетку вместимостью 5 см, к верхнему концу которой припаян трехходовой кран. На верхний отвод тройника надета резиновая груша. Раствор Б хранят в колбе вместимостью 100 см', откуда набирают в пипетку, а из пипетки спускают в эбулиостат.
В зависимости от массовой доли PB в растворе и интенсивности его окраски применяют два варианта титрования: для светлых растворов, содержащих 0,13...0,05% PB1 используют прямое титрование раствором сахара; для темных растворов или растворов, содержащих меньше 0,05% PB1 применяют дотитровывание раствором глюкозы.
Методика анализа по первому варианту. В колбу прибора наливают воду и ставят прибор на электрическую плитку. Для равномерного образования пара на дно колбы помещают кусочки пористого стекла или шамота. Когда вода закипит, вынимают из колбы эбулиостат и наливают в него из дозаторов сначала 5 см3 раствора А, а затем 5 см3 раствора Б. Осторожно перемешивают растворы в эбулиостате и медленно вставляют эбулиостат вместе с пробкой в колбу.
Затем верхнее отверстие эбулиостата закрывают пробкой с бюреткой, в которую наливают исследуемый раствор сахара. Регулируют выход пара через отводную трубку таким образом, чтобы жидкость в эбулиостате нагрелась до температуры кипения воды примерно за 0,5 мин. Как только пар начнет проходить через медно-щелочной раствор, начинают титрование раствором сахара.
Сначала проводят ориентировочное определение. Раствор сахара из бюретки добавляют по каплям со скоростью одна капля за 1...2 с до перехода окраски раствора из синей в желтую и замечают объем раствора сахара, израсходованного на титрование. Затем проводят окончательное определение. В эбулиостат заливают новую порцию медно-щелочного раствора, подготавливают эбулиостат к титрованию, как указано выше, и начинают титрование. Вначале сразу прибавляют 80...90% объема раствора сахара, израсходованного при ориентировочном определении, и через 2 мин с начала пробулькивания пара через жидкость в эбулиостате дотитровывают, прибавляя раствор сахара со скоростью одна капля за 6...7 с, до перехода синей окраски в желтую от одной капли сахарного раствора.
По бюретке определяют объем раствора сахара, израсходованного на титрование 10 CMi медно-щелочного раствора.
Массовую долю PB1 %, в гидролизате легкогидролизуемых полисахаридов или в разбавленном нейтрализованном гидролизате трудногидролизуемых полисахаридов г, и ст соответственно рассчитывают по формуле
![]() 1
1
где T1 - титр медно-щелочного раствора по глюкозе для первого варианта, мг; х - объем гидролизата, израсходованный на титрование 10 см1 медно-щелочного раствора.
Методика анализа по второму варианту. Подготавливают прибор к работе, как и в первом варианте, но в эбулиостат кроме 5 см1 раствора А и 5 см3 раствора Б, добавляют, отмеривая пипеткой, определенный объем раствора сахара. В случае темного раствора, содержащего более 0,05% PB, этот объем должен составлять 1...2 см3 , а в случае раствора с массовой долей PB менее 0,05% - 5 см3 . Затем к жидкости в эбулиостате добавляют из бюретки такой объем раствора глюкозы концентрацией около 0,1 г/100 см3 , чтобы на дотитровывание после 2 мин кипячения потребовалось бы не более 1 см3 этого раствора. Добавляемый объем определяют предварительным анализом. Концентрация добавляемого раствора глюкозы должна быть установлена точно; удобнее всего пользоваться раствором глюкозы, приготовленным для установки титра медно-щелочного раствора,
После перемешивания жидкости эбулиостат с пробкой медленно вставляют в колбу, закрывают его пробкой с бюреткой, в которую налит раствор глюкозы. Через 2 мин с начала пробулькивания пара через жидкость в эбулиостате проводят дотитровывание раствором глюкозы, добавляя его со скоростью одна капля за 6...7 с до перехода окраски в желтую от одной капли раствора глюкозы, израсходованного на дотитровывание.
Массовую долю PB,%, в гидролизате легкогидролизуемых полисахаридов или в разбавленном нейтрализованном гидролизате трудногидролизуемых полисахаридов сл и сг соответственно рассчитывает по формуле
![]()
где T2 - титр медно-щелочного раствора по глюкозе для второго варианта, мг; с.
При фотоколориметрическом методе для обнаружения бесцветных Сахаров на бумаге хроматограмму проявляют - обрабатывают раствором проявителя, дающего с сахарами окрашенные соединения. Количества разделенных моносахаридов приближенно можно определить непосредственно на хроматограмме прямым измерением интенсивности окраски пятен. Однако из-за нестабильности окраски и существенного влияния условий обработки хроматограмм точность этого метода невелика: относительная ошибка измерений достигает 10...15%. Более точным способом является извлечение окрашенных продуктов из хроматограммы - э л ю и с о в а н и е с последующим фотометрированием полученных растворов - злюатов на фотозлектроколориметре или спектрофотометре.
Для разделения Сахаров применяют специальную хроматографическую бумагу № 1 или Ne 2, которую нарезают полосками. Скорость разделения больше на полоске бумаги с продольным расположением волокон, чем на полоске с поперечным расположением. Иногда для лучшего разделения Сахаров необходимо обеспечить более медленное движение растворителя. С этой целью бумагу нарезают в направлении, перпендикулярном направлению волокон.
Хроматографирование осуществляют нисходящим или восходящим потоками жидкости. В первом случае бумагу подвешивают, укрепляя верхний конец в сосуде с растворителем. Во втором случае бумагу помещают в растворитель нижним концом. Когда сахара трудно разделяются, рекомендуется повторное хроматографирование.
Растворитель должен обеспечивать максимальное различие в значениях Rt и сам должен быть высокоподвижен. При разделении Сахаров используют, например, следующие системы растворителей: этилацетат - пиридин - вода.,; н-бутанол - уксусная кислота - вода; н-бутанол - ацетон - вода, и др. Разделение Сахаров зависит от состава растворителя.
Для повышения эффективности разделения растворителю дают возможность стекать до тех пор, пока сахара не разделятся по всей длине хроматограммы. При этом вычислить значения R1 невозможно, так как неизвестен путь, пройденный фронтом растворителя. В этом случае подвижность определяют косвенным способом, сравнивая пути, пройденные разделяемыми веществами, с перемещением пятна вещества с известным Rj . В химии углеводов часто используют ксилозу и определяют коэффициент подвижности Rkc как отношение расстояния, пройденного сахаром, к расстоянию, пройденному в этих же условиях ксилозой. Кроме природы сахара и состава растворителя на коэффициент подвижности влияют качество бумаги и температура среды.
В качестве проявителей применяют разнообразные реагенты в зависимости, от состава разделяемых смесей. Для проявления хроматограмм гидролизатов растительных тканей, содержащих редуцирующие сахара, одним из лучших проявителей считают раствор анилинфталата в этаноле. При проявлении анилинфталатом пентозы в зависимости от количества дают окраску от розовой до темно-красной, гексозы - зеленовато-коричневую, рамноза - светло-коричневую. Для элюирования окрашенных соединений из хроматограммы используют ледяную уксусную кислоту или раствор соляной кислоты в этаноле.
Методика анализа. Разделение Сахаров проводят нисходящим способом при непрерывном протекании растворителя по бумаге.
Подготовка гидролизата. Концентрация PB в гидролизате, подготовленном для хроматографирования, должна находиться в пределах 1...3%. При необходимости гидролизат упаривают. Упаривание проводят в вакуум-сушильном шкафу при температуре 50...60°С или при нагревании на водяной бане в фарфоровой чашке или в стеклянном стакане. Гидролизат легкогидролизуемых полисахаридов предварительно нейтрализуют концентрированным раствором гидроксида натрия до слабощелочной реакции и подкисляют концентрированной уксусной кислотой до рН 5, затем упаривают и фильтруют через простую конусообразную воронку с бумажным фильтром. Гидролизат трудногидролизуемых полисахаридов нейтрализуют карбонатом бария и отфильтровывают осадок сульфата бария. Полученный фильтрат сразу подвергают хроматографированию. При необходимости производят упаривание после подкисления уксусной кислотой до рН 5 и снова фильтруют. Кратность упаривания точно определяют и учитывают при расчете масс моносахаридов.
Приготовление растворителя. Для приготовления смеси этилацетат - пиридин - вода 150 см3 этилацетата и 30 см3 пиридина хорошо взбалтывают в делительной воронке вместимостью 500 см3 , затем прибавляют к смеси 150 см3 дистиллированной воды и снова хорошо взбалтывают в течение 2...3 мин. После этого смесь оставляют для расслаивания. Когда верхний слой жидкости станет прозрачным, нижний слой сливают, а верхний используют для хроматографирования. Подобным образом готовят и другие составы растворителей, смешивая органические растворители и воду в соответствующих пропорциях.
Хроматографическое разделение. Установка для хроматографирования представляет собой широкий цилиндр с притертой крышкой. Внутрь цилиндра помещают малый цилиндр, на который ставят чашку с растворителем.
Нарезают 10...12 полосок хроматографической бумаги размером 3ч45 см. При хроматографировании нижний конец полоски может свободно свисать либо его вырезают в виде "язычка", который вставляют в горлышко колбочки-приемника. На расстоянии 10...15 см от другого конца полоски отмечают точку старта, на которую наносят определенное количество подготовленного гидролизата. Все отметки, на бумажной полоске делают только графитовым карандашом.
Пробу гидролизата наносят микропипеткой со специальным приспособлением, состоящим из резиновой груши в металлическом кожухе и микровинта с шариком. Если упаривание гидролизата нежелательно, то на бумагу наносят несколько раз в одно и то же место необходимый объем раствора сахара, который рассчитывают исходя из массовой доли PB. После нанесения каждой порции раствора пятно хорошо просушивают. Диаметр нанесенного пятна не должен превышать 5...6 мм.
Полоски устанавливают в строго вертикальном положении. Конец полоски, на который нанесена проба, загибают в чашку. Пятно пробы должно находиться снаружи чашки ниже ее края не менее чем на 2 см. Второй конец вставляют в колбочку-приемник. Для лучшей идентификации Сахаров в установку помещают также полоску бумаги с нанесенным раствором смеси чистых моносахаридов: галактозы, глюкозы, маннозы, арабинозы, ксилозы. В чашку наливают растворитель и установку герметично закрывают крышкой.
Хроматографирование проводят при температуре 18...22°С, помещая установку в специальную комнату или шкаф. Разделение осуществляется за 24...90 ч. Хроматографирование считают законченным тогда, когда на проявленной хроматограмме расстояния между пятнами будут не менее 3 мм.
После окончания разделения Сахаров хроматограммы сушат на воздухе в течение примерно 4 ч до полного удаления растворителя. При сушке хроматограммы перекидывают через стеклянную палочку, укрепленную на двух штативах.
Проявление хроматограмм. Проявителем служит раствор, содержащий 1,66 г фталевой кислоты и 0,92 см3 свежеперегнанного анилина в 100 см3 этанола.
Сухие хроматограммы смачивают равномерно проявителем кратковременным погружением в раствор, отжимают между листами фильтровальной бумаги и немного подсушивают на воздухе. Затем хроматограммы помещают в сушильный шкаф, предварительно нагретый до нужной температуры. m Несколько хроматограмм одновременно кладут на ребро так, чтобы они не касались друг друга и стенок шкафа. Проявление ведут при температуре 100...105°С в течение 5 мин. Аналогичным образом проявляют полоски бумаги с нанесенным раствором смеси чистых Сахаров. При установлении природы Сахаров руководствуются цветом пятен, их относительным расположением и расположением пятен чистых Сахаров.
Фотоколориметрическое определение Сахаров. Из проявленной хроматограммы вырезают участки, соответствующие отдельным сахарам, и разрезают их на узкие полоски. Помещают кусочки хроматограмм в пробирки с пришлифованными пробками и заливают 8 см3 раствора HCI в этаноле. Элюирование ведут в течение 1 ч при комнатной температуре, поместив пробирки в темноту. Через каждые 5...10 мин пробирки энергично встряхивают.
Элюаты сливают в подготовленные сухие пробирки и фото-метрируют - измеряют оптическую плотность на фотоэлектрическом колориметре при синем светофильтре в кювете толщиной 10 мм или же на спектрофотометре при длине волны 420 нм.
Массу каждого моносахарида определяют по соответствующим градуировочным графикам.
Зная массу сахара в пробе гидролизата, нанесенной на хроматограмме, объем этой пробы, кратность упаривания и общий объем гидролизата, рассчитывают массовую долю, % к абсолютно сухой древесине, соответствующих полисахаридов Р - глюканов, маннанов, галактанов, ксиланов, арабинанов
![]()
где Ь - масса моносахарида в пробе гидролизата, мкг; х - объем пробы гидролизата, взятый на хроматографирование, см3 ; V - общий объем гидролизата, V=500 - или 200 см3 ; k - коэффициент пересчета моносахаридов на полисахариды, £ = 0,90 для гексозанов и 0,88 для пентозанов; з - кратность упаривания; g - масса абсолютно сухой навески древесины, г.
Построение градуировочных графиков. Градуировочный график для каждого моносахарида представляет собой зависимость оптической плотности элюата от массы сахара на хроматограмме. Для построения графиков готовят точно 1% -ные растворы чистых моносахаридов. На полоску хроматографической бумаги на расстоянии 3...4 см друг от друга наносят разные количества 1% -ного раствора сахара: 0,01; 0,02; 0,03; 0,04 и 0,05 см3 . После высыхания пятен полоску проявляют раствором анилинфталата в этаноле в точно таких же условиях, как и при анализе гидролизатов. Проводят элюирование и фотометрирование, как описано выше. На основании средних данных нескольких параллельных определений для каждого моносахарида строят градуировочный график. Иногда градуировочный график для глюкозы используют для всех гексоз, а график для ксилозы - для всех пентоз. Последний способ менее точен.
6. Анализ гидролизатов методом газожидкостной хроматографии
В гидролизатах происходит мутаротация моносахаридов, т.е. образование равновесной смеси таутомерных форм: б-, в-пиранозных, б-, в-фуранозных и открытой. Для глюкозы это иллюстрирует следующая схема:
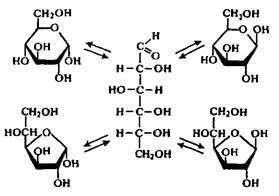
Существование таутомерных форм затрудняет определение состава моносахаридов в гидролизате.
Моносахариды нелетучи и непосредственное разделение их смесей методом газожидкостной хроматографии невозможно. Сахара могут быть хроматографически разделены в виде летучих производных - простых и сложных эфиров. Широко распространен метод анализа в виде триметилсилильных производных моносахаридов. Последние являются летучими гликозидами простых О-триметилсилиловых эфиров углеводов, образующихся в результате полного замещения гидроксильных групп моносахаридов при их силировании.
В качестве силирующего реагента моносахаридов используют смесь триметилхлорсилана и гексаметилдисилазана в среде пиридина.
В качестве примера приводится схема реакции силирования для галактозы
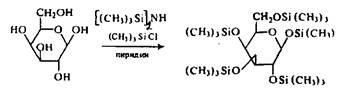
Подобным образом синтезируются TMС-производные других моносахаридов. При этом устанавливается новая равновесная смесь, в которой преобладают пиранозные формы. Соотношение таутомеров в зависимости от условий колеблется. Состав ТМС-производных таутомерных форм индивидуальных моносахаридов приведен ниже.
Для уменьшения числа пиков на хроматограмме и упрощения ее обработки моносахариды восстанавливают до соответствующих многоатомных спиртов с последующим их превращением в ацетаты. Каждый полиол дает на хроматограмме один пик, поскольку таутомерия у полиолов невозможна.
При анализе ТМС-производных методом газожидкостной хроматографии инертный газ служит подвижной фазой, неподвижной фазой является жидкость, нанесенная тонким слоем на твердый носитель. В качестве жидкой фазы используют различные силиконовые масла и полиэфиры. Наиболее селективными фазами из вырабатываемых отечественной промышленностью являются неполярное метилхлорсиликоновое масло и полярный политриэтиленгликольсебацинат. В качестве твердого носителя применяют адсорбенты порохром-3 или хромат N.
Для хроматографического разделения смеси летучих производных моносахаридов используют газовые хроматографы, состоящие из колонки с термостатом, в которой происходит разделение смеси, пламенно-ионизационного детектора, дающего сигнал прямо пропорциональный количеству анализируемого вещества, блоков управления и самопишущего прибора. Выход ТМС-производных, регистрирующийся самопишущим прибором в виде пика на диаграммной ленте, соответствует времени удерживания tR - времени, прошедшего с момента введения пробы в колонку до момента появления компонента смеси в детекторе.
В соответствии с tR на хроматограмме в определенном порядке появляются пики индивидуальных соединений. Xpoматограмма - совокупность пиков, соответствующая порядку выхода и массе индивидуальных компонентов смеси. Порядок выхода при хроматографическом разделении ТМС-производных зависит от жидкой фазы. Его устанавливают предварительно для данной фазы с использованием модельных ТМС-производных чистых моносахаридов по их времени удерживания. На рис. приведена в качестве примера хроматограмма модельной смеси, разделенной на колонке с метилхлорфенил-силиконовым маслом в качестве жидкой фазы.
Существует несколько методов количественного определения состава смеси по полученным хроматограммам. По первому методу определяют площади пиков всех таутомеров каждого моносахарида расчетом площади треугольной аппроксимацией пика - умножением основания пика на половину высоты. При частичном наложении пиков друг на друга и невозможности прямого измерения основания площадь пика определяют умножением его высоты на ширину, измеренную на половине высоты. Возможно также определять долю каждого пика его переносом на бумагу и взвешиванием вырезанного из нее пика на аналитических весах, относя найденную массу к массе всех анализируемых пиков. Результат выражают в процентах, используя метод нормировки площадей.
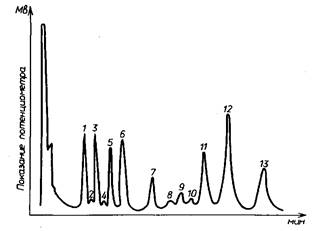
Хроматограмма модельной смеси TMC производных моносахаридов
По второму методу количественное определение массовой доли конкретного моносахарида в смеси производят по площади пика одного из его таутомеров, исходя из условия, что при синтезе ТМС-производных в строго постоянных условиях соотношение таутомеров сохраняется постоянным. Это позволяет для расчета выбрать наиболее разрешенный пик, называемый характеристическим. В качестве таких пиков обычно используют пики ТМС-производных в-Ж,-арабинопиранозы, в-П-кси-лопиранозы, a-D-маннопиранозы, a-D-галактопиранозы и в-D-глюкопиранозы. Площади пиков остальных таутомеров непосредственно не измеряют, а находят расчетом. С учетом доли характеристического пика вычисляют общую площадь пиков всех таутомеров данного моносахарида. Количественное соотношение таутомерных форм необходимо устанавливать для данных условий хроматографирования и при. их изменении определение следует проводить вновь. Долю конкретного моносахарида находят отнесением площади всех пиков данного моносахарида к общей площади всех пиков.
Количественное определение массы индивидуальных моносахаридов производят с использованием внутреннего стандарта. Масса сорбита, добавляемая в анализируемую смесь, должна обеспечить его концентрацию в растворе, примерно соответствующую концентрации индивидуальных моносахаридов. Расчет ведут по градуировочному графику. График получают, по данным хроматографирования эталонных растворов, содержащих индивидуальный моносахарид и сорбит. Градуировочный график строят в координатах: по оси абсцисс - масса чистого моносахарида в эталонном растворе, по оси ординат - отношение площади характеристического пика этого же моносахарида к площади пика внутреннего стандарта. График имеет линейную форму. Прямую линию проводят с использованием метода наименьших квадратов.
Относительная ошибка определений Сахаров в гидролизате зависит от их массовой доли в жализируемой смеси и составляет 1...5%. Общее время анализа при серийном проведении определений около 2 ч, не считая времени на получение градуировочных графиков.
Методика анализа. Анализ гидролизатов методом газожидкостной хроматографии включает подготовку гидролизатов к анализу, синтез ТМС-производных моносахаридов, хроматографическое разделение этих производных и обработку хроматограмм.
Подготовка гидролизатов к анализу. Гидролизаты, полученные при обработке древесины концентрированной серной кислотой, нейтрализуют карбонатом бария до рН 5. Раствор отфильтровывают через конусообразную стеклянную воронку с бумажным фильтром от осадка сульфата бария. Гидролизаты, полученные при обработке древесины соляной кислотой, нейтрализуют карбонатом натрия до рН 5. В полученных гидролизатах определяют массовую долю PB. При необходимости гидролизат упаривают в вакуум-сушильном шкафу при температуре 40°С.
Синтез ТМС-производных моносахаридов. В круглодонную колбу вместимостью 50 см3 вносят пипеткой пробу нейтрализованного и упаренного гидролизата, содержащего около 20 мг PB.
Работу выполняют в двух вариантах - с внутренним стандартом или без него.
Раствор сорбита вносят в колбу пипеткой в объеме, соответствующем его массе, равной 2...5 мг. Колбу присоединяют к ротационному вакуумному испарителю и выпаривают раствор при температуре 40...42°С и остаточном давлении 0,5...1 кПа. Выпаривание производят досуха. Для удаления следов воды к упаренной пробе добавляют 2...3 раза по 1 см3 спиртотолуольной смеси, которую также удаляют упариванием. Затем сухой остаток в этой же колбе растворяют в 2 см3 свежеперегнанного сухого пиридина, добавляют 0,6 см3 гексаметилдисилазана и 0,3 см3 триметилхлорсилана. Колбу закрывают пробкой, энергично встряхивают 30 с и при комнатной температуре выдерживают реакционную смесь в течение 10 мин.
Если анализируемая проба плохо растворяется, колбу нагревают на водяной бане при температуре 75...85°С в течение 2...3 мин. Затем проводят упаривание пиридина из реакционной смеси на ротационном испарителе при температуре 40°С и остаточном давлении 0,5...1 кПа в течение 10 мин. Остаток растворяют в этой же колбе в 2 см3 "-гексана. В хорошо закрытых колбах ТМС-производные устойчивы в течение нескольких недель.
Массовую долю каждого моносахарида в гидролизате, %, рассчитывают по формуле
![]()
где т, - масса моносахарида в пробе гидролизата, мг; х - объем пробы гидролизата, см3 .
По полученным значениям с, Для гидролизатов легко - и трудногидролизуемых полисахаридов рассчитывают массовые доли соответствующих полисахаридов в процентах по отношению к абсолютно сухой древесине.
По второму варианту рассчитывают долю каждого моносахарида в процентах от их обшей массы по методу нормировки площадей. Вычисляют сумму площадей всех пиков S и площади пиков каждого моносахарида S4 .
Массовую долю каждого моносахарида, % к их общей массе, рассчитывают по формуле
![]()
Определение времени удерживания моносахаридов и построение градуировочных графиков. По первому варианту анализа готовят эталонные растворы определенной концентрации чистых моносахаридов и внутреннего стандарта - сорбита. Для каждого моносахарида готовят пять эталонных растворов концентрацией от 2 до 10 мг/см3 . Концентрация раствора сорбита постоянна - 4 мг/см3 . Из каждого раствора отбирают три пробы, синтезируют TMC-производные и хроматографируют, как указано выше. На каждой из трех хроматограмм для каждого пика фиксируют значение tR , вычисляют площадь характеристического пика таутомера моносахарида S1 и площадь пика сорбита Scl . Таким образом находят S,/Scr для всех пяти эталонных растворов. По средним значениям результатов трех параллельных анализов для каждого из пяти эталонных растворов строят графики зависимости отношений SJScr от массы моносахаридов, пользуясь методом наименьших квадратов.
Время удерживания для каждого моносахарида находят по шкале времени хроматограмм.