| Похожие рефераты | Скачать .docx | Скачать .pdf |
Реферат: Наследственные заболевания обмена веществ
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ОБМЕНА ВЕЩЕСТВ
К настоящему времени известно более 2500 наследственных ферментопатий, 20 классов наследственных болезней обмена, но только для части из них (для 300) установлен точный уровень метаболического блока и характер ферментативного дефекта. До сих пор большая часть этих заболеваний не диагностируется или выявляется поздно, когда происшедшие нарушения носят необратимый характер. Одна из трудностей ранней диагностики заключается в том, что в период новорожденности у этих детей нет специфических расстройств, а поздние проявления фенотипически схожи с заболеваниями ненаследственного генеза. Вторая особенность состоит в том, что для наследственных заболеваний обмена веществ характерен клинический полиморфизм, обусловленный генетической гетерогенностью. Это объясняется наличием множественных изоаллельных мутаций и возможностью возникновения мутаций в разных генах.
Клинические проявления наследственных болезней обмена веществ во многом определяются поражением нервной системы (особенно при нарушениях обмена аминокислот, липидов и кислых гликозамино-гликанов), что в свою очередь, усиливает имеющиеся нарушения и усугубляет тяжесть клинических проявлений заболевания. Для диагностики наследственных болезней важен анализ неврологических симптомов, особенно на ранних стадиях развития, и разграничение их от фенокопий- заболеваний ненаследственной природы со сходной клинической картиной. Важное практическое значение имеет выявление гетерозиготных носителей мутантного гена при наследственных болезнях. Например, при частоте фенилкетонурии 1:10 000, мутантный ген встречаетсяв популяции 1:50. Гетерозиготное состояние может клинически не обнаруживаться, поэтому выявление умеренно выраженных изменений метаболизма является единственным указанием на наличие мутантного гена.
Наследственные болезни обмена аминокислот
Роль аминокислот для организма человека чрезвычайно велика. Аминокислоты являются основными структурными элементами белков, необходимы для синтеза иммуноглобулинов, гормонов, служат источником энергии. Каждый фермент или белок имеет специфические свойства и функции, которые определяют и регулируют сложные обменные процессы и развитие организма.
Часть аминокислот не может синтезироваться в организме человека. Это незаменимые аминокислоты: триптофан, фенилаланин, метионин, лизин, лейцин, изолейцин, валин и треонин. В детском возрасте к их числу относится гистидин, т.к. организм ребенка не может синтезировать эту аминокислоту в необходимых для нормального роста количествах. Клетки растущих тканей содержат аминокислоты в высоких концентрациях, что является свидетельством высокой интенсивности процессов транспорта аминокислот через клеточные мембраны.
Для обеспечения нормального роста и развития важно не только количество поступающих аминокислот, но и их соотношение. При избытке или недостатке аминокислот развиваются явления аминокислотного дисбаланса. Например, избыток лейцина в пище тормозит рост организма, метионина- вызывает токсическое поражение нервной системы, цистина- способствует развитию жировой инфильтрации печени.
Таким образом, нарушения метаболизма аминокислот приводят к нарушению нормального функционирования организма человека.
Наследственные нарушения обмена аминокислот
(Ю.И. Барашнев, Ю.Е. Вельтищев, 1978 г.)
1. Наследственные нарушения обмена аминокислот, сопровождающиеся увеличением их концентрации в крови и моче: фенилкетонурия, гистидинемия, триптофанурия, болезнь "кленового сиропа", орнитинемия, цитруллинемия и др. Наследование, в основном, по аутосомно-рецессивному типу. В основе развития заболеваний лежит нарушение синтеза или структуры тех или иных ферментов.
2. Наследственные нарушения обмена аминокислот, сопровождающиеся увеличением их выделения с мочой без изменения уровня в крови: гомоцистинурия, гипофосфатазия, аргиносукцинатацидурия и др. При данных энзимопатиях нарушено обратное всасывание в почках, что приводит к увеличению их содержания в моче.
3. Наследственные нарушения систем транспорта аминокислот: цистинурия, триптофанурия, болезнь Гартнепа и др. К этой группе относятся энзимопатии, развитие которых обусловлено снижением реабсорбции аминокислот в почках и кишечнике.
4. Вторичные гипераминоцидурии: синдром Фанкони, фруктоземия, галактоземия, болезнь Вильсона-Коновалова и др. При данных состояниях возникает вторичная генерализованная гипераминоацидурия в результате вторичных тубулярных нарушений.
Фенилкетонурия (ФКУ)
Впервые описана в 1934 г. Folling под названием "фенилпировиноградная имбецильность". Тип наследования - аутосомнорецессивный. Частота заболевания составляет 1:10000- 1:20000 новорожденных. Пренатальный диагноз возможен при использовании генетических зондов и биопсии ворсин хориона.
К развитию классической клинической картины при ФКУ приводит недостаточность фенилаланингидроксилазы и недостаточность редуктазы дигидроптерина- 2-го фермента, обеспечивающего гидроксилирование фенилаланина. Их недостаток приводит к накоплению фенилаланина (ФА) в жидких средах организма (схема 1). Как известно, ФА относится к незаменимым аминокислотам. Поступающий с продуктами питания и не используемый для синтеза белка, он распадается по тирозиновому пути. При ФКУ наблюдается ограничение превращения ФА в тирозин и, соответственно, ускорение его превращения в фенилпировиноградную кислоту и другие кетоновые кислоты.
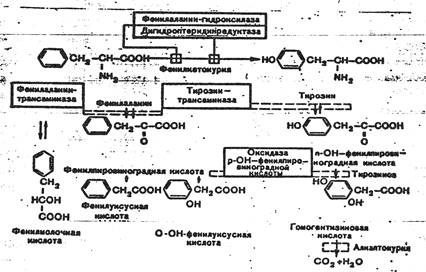
Схема 1. Варианты нарушений метаболизма фенилаланина.
Существование различных клинико-биохимических вариантов ФКУ объясняется тем, что фенилаланингидроксилаза является частью мультиферментной системы.
Различают следующие формы ФКУ:
1. Классическая
2. Скрытая.
3. Атипичная.
Развитие атипичных и скрытых форм ФКУ связывают с недостаточностью фенилаланинтрансаминазы, тирозинтрансаминазы и оксидазы парагидроксифенилпировиноградной кислоты. Атипичная ФКУ обычно не сопровождается поражением нервной системы в результате позднего развития ферментативного дефекта.
У женщин с фенилкетонурией возможно рождение детей с микроцефалией, задержкой умственного развития, нарушениями развития мочевыделительной системы, поэтому необходимо назначение диетотерапии во время беременности.
Клинические симптомы у больных ФКУ
При рождении ребенок с фенилкетонурией выглядит здоровым. Заболевание у этих детей проявляется на первом году жизни.
1. Интеллектуальный дефект. Нелеченный ребенок теряет около 50 баллов IQ к концу 1-го года жизни. У больных не выявляется зависимости между уровнем ФА и степенью интеллектуального дефекта.
2. Судорожный синдром (4 50%), экзема, гипопигментация.
3. Нарушение координации движения.
4. Задержка развития статических и двигательных функций.
5. Поражение пирамидных путей и стриопаллидарной системы. Клинические проявления классической ФКУ редко встречаются в странах, в которых действует программа неонатального скрининга на это заболевание.
У детей с фенилкетонурией наблюдается повышенный уровень в моче метаболитов ФА. Увеличение в физиологических жидкостях содержания ФА и недоокисленных продуктов его метаболизма приводит к поражению нервной системы. Определенная роль в этих нарушениях принадлежит дисбалансу аминокислот (дефицит тирозина, который в норме активно участвует в построении белкового компонента миелина). Демиелинизация является характерным патоморфологическим признаком фенилкетонурии. Нарушение соотношения аминокислот в
крови приводит к нарушению уровня свободных аминокислот в головном мозге, что вызывает слабоумие, гиперкинезы и другие неврологические симптомы.
Пирамидные симптомы обусловлены нарушением процессов миелинизации. Избирательный характер поражения нервной системы объясняется особенностями миелинизации- поражаются наиболее молодые в филогенетическом отношении отделы, выполняющие сложные и дифференцированные функции. С недостаточным образованием меланина из тирозина связывают голубой цвет глаз, светлую кожу. Запах "плесени" ("мышиный", "волчий") объясняется наличием фенилуксусной кислоты в моче. Кожные проявления (экссудативный диатез, экземы) связаны с выделением аномальных метаболитов. Недостаточность образования адренергических гормонов из тирозина приводит к артериальной гипотонии.
Необходимо отметить, что при ФКУ в патологический процесс вовлекается печень, но характер морфологических расстройств не является специфичным: выявляются признаки тканевой гипоксии, нарушения окислительной и белоксинтезирующей функции, перегрузка липидами. Наряду с этим наблюдаются компенсаторно-приспособительные изменения: высокое содержание гликогена, гиперплазия митохондрий. Генерализованную гипераминоацидемию при ФКУ можно объяснить вторичным нарушением метаболизма аминокислот в связи с повреждением гепатоцитов, т.к. многие ферменты, участвующие в аминокислотном обмене, локализуются в печени.
У нелеченых больных с классической ФКУ наблюдается значительное снижение концентрации катехоламинов, серотонина и их производных в моче, крови, ликворе. Поэтому в комплексном лечении ФКУ необходима промедиаторная коррекция, так как парциальный интеллектуальный дефект может быть связан с нейромедиаторными нарушениями.
Критерии диагностики классической формы фенилкетонурии:
1. Уровень ФА в плазме выше 240 ммоль/л.
2. Вторичный дефицит тирозина.
3. Повышенный уровень в моче метаболитов ФА.
4. Сниженная толерантность к полученному внутрь ФА.
Методы диагностики фенилкетонурии:
1. Проба Феллинга с FeCl3 - при положительном анализе появляется сине-зеленое окрашивание мочи.
2. В крови выявление избытка фенилаланина возможно с помощью бактериального экспресс- теста Гольдфарба или теста Гатри (т.к. в течение первых дней жизни фенилпировиноградная кислота в моче может отсутствовать).
При ФКУ проводится лечение диетой с ограниченным содержанием ФА (главным образом назначают овощные блюда, мед, фрукты). Такие продукты, как молоко, молочные изделия, яйца, рыба, должны быть полностью исключены в период пребывания больных с ФКУ на острой диете. Назначаются специальные препараты (цимогран, лофеналак) и витамины.
Оптимальные сроки обследования новорожденных- 6-14 день жизни, начало терапии - не позднее 21 дня жизни. Необходимо помнить, что проведение исследования в первые сутки не исключает ложноположительных или ложноотрицательных результатов ( повторное исследование проводят до 21 дня жизни ). Эффективность лечения оценивается по интеллектуальному уровню развития пациента. Необходимо отметить, что лечение, начатое после года не нормализует интеллект полностью (возможно, это связано с развитием необратимых изменений в мозге).
ГОМОЦИСТИНУРИЯ
Заболевание впервые описано в 1962 г. Carson и Neil. Наследуется по аутосомно-рецессивному типу. Частота гомоцистинурии составляет 1:200 000 новорожденных.
В основе заболевания лежит отсутствие или снижение активности фермента цистатионинсинтетазы, что ведет к нарушению обмена метионина. Кофактором цистатионсинтетазы является витамин В6 . Поэтому наблюдается пиридоксинчувствительная и пиридоксинрезистентная формы. У родителей и родственников больных часто обнаруживают шизофрению. Рядом авторов отмечается фенотипическое сходство с болезнью Марфана. Однако, при гомоцистинурии в отличие от болезни Марфана более выражены изменения нервной системы, снижение интеллекта и судорожный синдром.
Формы гомоцистинурии:
1. Классическая.
2. Связанная с дефицитом утилизации витамина В6 .
3. Обусловленная нарушением метаболизма фолиевой кислоты.
Клинически дети, больные классической формой, при рождении выглядят здоровыми. Возможны лишь задержка роста и развития. Диагноз обычно устанавливается после 3-х лет, когда выявляется подвывих хрусталика. Основной рентгенологический признак- генерализованный остеопороз. При гомоцистинурии наблюдается клинический полиморфизм. Однако, можно выделить своеобразный комплекс признаков (таблица 1).
Таблица 1. Симптомокомплекс гомоцистинурии
( Ю.И.Барашнев, Ю.И.Вельтищев, 1978 г.)
| Симптомы | Фенотипические проявления |
| Изменение скелета | Укороченное туловище,удлиненные конечности, нарушенная осанка, "башенная" форма черепа, неправильный прикус и рост зубов,высокое небо, "крыловидные" лопатки, воронкообразная деформация грудной клетки, вальгусная деформация грудной клетки, вальгусная деформация нижних конечностей, плоскостопие, умеренный остеопороз |
| Изменение нервной системы | Сниженный интеллект,патологический характер ЭЭГ, спастическая походка |
| Нарушение зрения | Подвывих хрусталиков, вторичная глаукома, изменения глазного дна |
| Сердечно- сосудистые расстройства | Нарушение обменных процессов в миокарде (по данным ЭКГ) |
| Внешние признаки | Светлые, мягкие,вьющиеся крупными завитками волосы, голубой цвет радужной оболочки |
Гомоцистинурия ведет к поражению соединительной ткани за счет усиления синтеза сульфатированных протеогликанов с последующей дегенерацией эластических элементов, депозицией коллагена и кальцификацией. Вовлечение в патологический процесс соединительной ткани приводит к костным деформациям, подвывиху хрусталика книзу. Генез подвывиха хрусталика следующий: дефицит цистина приводит к вовлечению в процесс соединительнотканных волокон цинновых связок. У больных с гомоцистинурией выявляются разнообразные поражения органа зрения: близорукость, изменения глазного дна, вторичная глаукома. Резкое падение остроты зрения приводит к инвализации больных.
При гомоцистинурии с мочой избыточно выводится гомоцистин в результате инактивации цистатионинсинтетазы в головном мозге и печени больных. Изменения ЦНС у детей с гомоцистинурией обнаруживаются в 50 % случаев. Происхождение интеллектуального дефекта при этом заболевании до сих пор не ясно. Отмечено сочетание гомоцистинурии с эпилепсией, шизофренией, задержкой интеллектуального развития. Высказываются предположения, что основной причиной умственного дефекта при гомоцистинурии являются сосудистые мозговые тромбы, в связи с чем образуется много мелких инфарктов мозга. При этом возникают некротические и дегенеративные изменения в мозговом стволе, зрительном бугре и коре.
У больных более старшего возраста (до 30 лет) наблюдается склонность к артериальным и венозным тромбозам, что связано с активацией гомоцистином фактора Хагемана, а также с оседанием гомоцистина из-за его низкой растворимости в патологически измененной интиме сосудов. Оба этих процесса могут создавать условия для образования тромбов.
Необходимо отметить, что гомоцистинурии свойственен прогредиентный характер течения патологического процесса. При рождении дети обычно не имеют каких-либо внешних дефектов. Но в результате наследственных нарушений постепенно начинают выявляться изменения в отдельных органах и системах. Существует зависимость клинической картины от возраста. Чем старше ребенок, тем больше органов и систем вовлекается в патологический процесс. Таким образом, гомоцистинурия - это заболевание всего организма. Диагностическим признаком является избыточная экскреция с мочей гомоцистина (необходима свежесобранная моча, т.к. гомоцистин разрушается при хранении).
ГИСТИДИНЕМИЯ
Заболевание впервые описано в 1961 г. Ghadimi с соавторами, а термин "гистидинемия" предложен Auerbach с соавт. в 1962 году.
Заболевание возникает в результате отсутствия или недостаточности активности фермента гистидазы. Наследуется аутосомно-рецессивно. Высказывается мнение об аутосомно-рецессивной передаче с неполной пенетрантностью, сцепленном с Х-хромосомой наследовании. Т.е., возможно, различные формы гистидинемии наследуются по разному. Для детей первого года жизни гистидин - незаменимая аминокислота. При недостатке гистидина в этом возрасте отмечается нарушение ретенции азота, выявляется дефицит массы тела, появляется шелушение кожи и экзематозные высыпания. Существует точка зрения о незаменимости гистидина и для здоровых взрослых (при недостатке наблюдаются признаки анемии с нарушением эритропоэза).
При гистидинемии происходит нарушение самого эффективного активного пути катаболизма- превращение гистидина в уроканиновую кислоту (этим путем в норме катаболизируется большая часть гистидина). В результате метаболического блока происходит накопление в крови и моче гистидина. Увеличение активности трансаминирования и усиленный перевод гистидина в имидазолпировиноградную, имидозолмолочную и имидозолуксусную кислоты является компенсаторно-приспособительной реакцией организма.
Гистидинемия отличается большой вариабельностью клинических проявлений: от тяжелой умственной отсталости до полного отсутствия каких-либо симптомов. Снижение интеллекта выявляется лишь у 50 % больных детей. Больные гистидинемией имеют светлый цвет волос, голубые глаза. На первый план у таких детей выступает поражение нервной системы: снижение интеллекта, нарушение речи, судороги. А у детей с нормальным интеллектом можно выявить особенности психики при гистидинемии: эмоциональную лабильность, агрессивность. Характер повреждения нервной системы при данном заболевании зависит от степени инактивации гистидидазы. Иногда гистидинемия сочетается с аномалиями развития, патологией почек, костной системы.
Для диагностики заболевания необходимо выявление повышенного уровня гистидина в плазме. Окончательный диагноз подтверждает определение гистидидазы в ороговевающем эпителии или печени.
Наследственные нарушения обмена триптофана
Как известно, триптофан является незаменимой аминокислотой.
Образующийся при расщеплении белков триптофан через кишечную стенку всасывается в кровь и используется организмом для синтеза белков. Остальная часть идет на синтез биологически активных веществ: никотинамида, серотонина, мелатонина и др. Таким образом, процессы метаболизма триптофана идут по трем основным направлениям: серотониновому, индольному и кинурениновому.
К наследственно обусловленным нарушениям обмена триптофана относится ряд клинических синдромов и заболеваний:
1. Болезнь Гартнепа.
2. Индиканурия.
3. Синдром Тада.
4. Синдром Прайса.
Болезнь Гартнепа
Впервые описана Baron с соавт. в 1956 году. Аутосомно- рецессивный тип наследования. При данном заболевании наблюдается генетическое изменение транспортной функции клеток слизистой оболочки кишечника и проксимальных отделов почечных канальцев. Это ведет к изолированному дефекту транспорта моноаминокарбоновых кислот. Нарушение кишечной абсорбции триптофана приводит к его бактериальному расщеплению в кишечнике до индола и индоксила. Генерализованная аминоацидурия обусловлена нарушением канальцевой реабсорбции.
Для болезни Гартнепа характерны кожная фоточуствительность, пеллагроподобный дерматит, мозжечковая атаксия с вовлечением в процесс пирамидных путей, нарушение функции желудочно-кишечного тракта. У некоторых детей выявляется умственная отсталость.
Индиканурия
Впервые описана в 1965 году Bickel.
В основе заболевания лежит нарушение всасывания триптофана в кишечнике с образованием избыточного количества индола, который всасывается, окисляется, сульфатируется и выделяется в виде индикана. Последний окисляется под влиянием воздуха до голубого индикана, окрашивающего пеленки в синий цвет (болезнь "голубых пеленок").
При индиканурии наблюдается гиперкальциемия, нефрокальциноз, периодическая гипертермия.
Синдром Тада
Данный синдром впервые описан в 1963 году Tada с соавт. под названием "триптофанурия с нанизмом". Аутосомно- рецессивный тип наследования. При синдроме Тада наблюдается недостаток фермента триптофанпирролазы, катализирующего превращение триптофана в кинуренин. Нарушения связаны с эндогенным дефицитом никотиновой кислоты и избытком индольных соединений. При синдроме Тада отмечается глубокая умственная отсталость, нанизм, мозжечковая атаксия.
Синдром Прайса
Впервые описан в 1967 г. Price с соавторами. Генетический дефект кинуренингидроксилазы. Наблюдается избыточное выделение с мочой кинуренина за счет блока фермента. Основное проявление синдрома Прайса - склеродермия.
Галактоземия
В 1908 году Reuss впервые отметил, что галактоземия относится к "врожденным ошибкам обмена". Изучению основного и побочного путей преобразования галактозы способствовали труды многих ученых.
Классическая галактоземия наследуется по аутосомно- рецессивному типу. По частоте занимает 2-е место после ФКУ. При этом заболевании наблюдается дефицит галактозо-1-фосфат-уридилтрансферазы. В эритроцитах гомозигот при классической галактоземии активность данного фермента почти не определяется, у гетерозигот составляет 50 % нормы.
Главный путь преобразования галактозы в глюкозу - это путь Лелуа. Первым этапом является фосфорилирование галактозы в печени, мозге, эритроцитах. В результате этой реакции образуется галактозо-1-фосфат, для дальнейшего превращения которого необходим специфический фермент галактозо-1-фосфат- уридилтрансфераза. При дефиците галактозо-1-фосфат - уридилтрансферазы невозможен дальнейший метаболизм галактозо-1-фосфата до УДФ- галактозы. Накопление галактозо-1-фосфата приводит к заболеванию, возможно, и во внутриутробном периоде. Кроме основного пути обмена галактозы (пути Лелуа), существуют обходные, дополнительные пути, которые необходимо учитывать в патологических условиях при дефиците галактозы.
Необходимо помнить, что из-за наличия большого числа фенокопий галактоземия не всегда распознается своевременно. Целый ряд болезней периода новорожденности клинически сходен с галактоземией: гемолитическая болезнь новорожденных, врожденный гепатит, атрезия желчных путей, энзимопатическая желтуха Криглера-Найяра и ряд других заболеваний.
При классической галактоземии наблюдается раннее появление симптомов заболевания: анорексия, рвота, непереносимость молока, задержка увеличения массы тела, катаракта, гепатомегалия, желтуха, отеки, геморрагические проявления, спленомегалия, поражение ЦНС. Задержка психомоторного развития выявляется рано и прогрессирует с возрастом (однако, умственная отсталость менее выражена, чем при ФКУ).
Тяжелые формы заболевания имеют яркую клиническую картину уже в первые дни жизни: расстройство пищеварения и резкая интоксикация (рвота, понос, гипотрофия), желтуха, которая имеет стойкий характер и наступает за счет увеличения концентрации прямого билирубина. Нередко печеночная желтуха сочетается с гемолитической за счет снижения осмотической резистентности эритроцитов. Необходимо отметить, что галактозо-1-фосфат является гепатотоксичным. Постепенно у больного прогрессируют признаки печеночной недостаточности. На 2-3 месяце жизни развивается асцит, спленомегалия, нарастают явления токсикоза и эксикоза. Повышенный уровень галактиола приводит к развитию катаракты (при тяжелой форме галактоземии катаракта может наблюдаться при рождении). Терминальная стадия болезни характеризуется печеночной недостаточностью, выраженной дистрофией и присоединением вторичных инфекций.
Диагностика заболевания основана на определении галактозы в крови и моче. Пренатальная диагностика- определение фермента в клетках амниотической жидкости. Тест на толерантность к галактозе не нужен и опасен, т.к. галактоза оказывает повреждающее действие.
Недостаточность лактазы
Под действием фермента лактазы в пищеварительном тракте человека происходит ферментативный гидролиз лактазы на глюкозу и галактозу. Существуют две разновидности фермента: "детского" и "взрослого" типа. Ген, контролирующий перевод лактазы "детского" типа во "взрослый", локализован на 2 хромосоме. В период до 5 лет происходит переключение синтеза фермента на "взрослый" тип. В некоторых случаях возможна персистенция "детской" лактазы во взрослом организме.
Формы лактазной недостаточности:
1. Алактазия- первичная наследственная лактазная недостаточность. Предполагаемый тип наследования аутосомно-рецессивный.
2. Транзиторная форма.
3. Гиполактазия взрослых.
4. Вторичная недостаточность лактазы.
Первичная недостаточность лактазы впервые описана A.Holzel в 1959г. Развивается тяжелая диспепсия, дегидратация, токсикоз и обезвоживание. Встречается редко, приводит к смерти в грудном возрасте.
Транзиторная недостаточность связана с незрелостью ферментов кишечника. В нормальных условиях активность кишечной лактазы возрастает на 24-40 неделе беременности. У недоношенных эта форма лактазной недостаточности проявляется поносом, рвотой, дегидратацией.
Взрослый тип чаще встречается у негров, индейцев и др. этнических групп. Среди русских- у 10-20 %. Клинически, как правило, не выявляется. Связан с персистенцией лактазы "детского" типа в результате мутации регуляторного гена.
Вторичная недостаточность лактазы может развиться при тяжелых расстройствах пищеварения, кишечных дисбактериозах, пищевой аллергии, лямблиозе, аскаридозе.
У детей с лактазной недостаточностью нарушается водно-солевое равновесие за счет повышенного выведения с мочой калия и кальция. Потеря кальция может привести к задержке оссификации хрящевой ткани у детей.
Лечение заключается в соблюдении безлактазной диеты. Эффективно применение фермента лактазы.
Врожденные нару шения обмена гликогена
Гликоген является важнейшим биополимером и содержится во всех клеточных органеллах. Этот полисахарид служит основным источником энергии и является резервом углеводов в организме.
В норме гликоген непрерывно подвергается обменным реакциям. В его синтезе и распаде участвует множество ферментов. Нарушение какого-либо звена в общей системе ферментов приводит либо к аномальному накоплению гликогена в клетках, либо к истощению его запасов. Врожденные нарушения в обмене гликогена могут возникать во всех органах (генерализованное заболевание) или в одном органе (чаще в мышцах или печени). Симптомы заболевания, вызванные одним и тем же ферментным дефицитом, могут проявляться в любом возрасте. В связи с этим различают инфантильную, юношескую и взрослую формы заболевания. Наиболее тяжелыми являются генерализованные заболевания инфантильной формы.
Необходимо отметить, что классификация гликогеновой болезни по формам заболевания, основанная на клинических данных, является рабочей классификацией клиницистов. В настоящее время принята классификация гликогенозов, предложенная Cori и основанная на ферментом дефекте заболевания (таблица 2).
Таблица 2 Болезни, вызванные нарушением обмена гликогена.
| Тип гликогеноза (название болезни) | Неактивный фермент | Органы, ткани и клетки, в которых найден дефект фермента | Клиническая форма болезни |
| I (болезнь Гирке) | Глюкозо-6 фосфатаза | Печень, почки,слизистая тонкого кишечника | Печеночная |
| II(болезнь Помпе) | Кислаяa глюкозидаза | Печень, почки,селезенка мышцы, лейкоциты, нерв ная ткань | Генерализованная |
| III(болезнь Кори) | Амило-1,6- глюкозидаза | Печень, мышцы, лейкоциты, эритроциты | Печеночная, мышечная |
| IV(болезньАндерсона) | Ветвящий фермент | Печень, мышцы, лейкоциты | Печеночная |
| V (болезньМак-Ардля) | Фосфорилаза мышц | Мышцы | Мышечная |
| VI(болезнь Херса) | Фосфорилаза печени | Печень | Печеночная |
| VII(болезньТомсона | Фосфоглюкомутаза | Мышцы, печень | Мышечная,Печеночная |
| VIII(болезнь Таруи) | Фосфофруктокиназа | Мышцы, эритроциты | Мышечная |
| IХ(болезньХага) | Киназа фосфорилазы b | Печень | Печеночная |
| Х | Протеинкиназа | Печень | Печеночная |
| ХI | Фосфогексоизомераза | Печень, эритроциты | Печеночная |
Гликогеноз I типа
Описан в 1952 г. C.Cori и G.Cori. Заболевание наследуется по аутосомно- рецессивному типу. Дефект фермента глюкозо-6-фосфатазы.
В зависимости от дефекта компонентов глюкозо-6-фосфатазной системы, различают три подтипа I типа гликогенозов: IА, IВ и IС.
IА - болезнь Гирке ( гепаторенальный гликогеноз) вызван отсутствием активности специфической глюкозо-6-фосфатазы в печени, слизистой кишечника, почках (скелетная и сердечная мускулатура, лейкоциты в процесс не вовлекаются, т.к. в нормальных условиях в этих тканях отсутствует глюкозо-6-фосфатаза).
IВ- связан с дефектом транспорта этого фермента в микросомальных мембранах гепатоцитов.
IС- транспортный дефект неорганического фосфата на уровне микросомальных мембран.
Глюкозо-6-фосфатазе принадлежит центральная роль в нормальном гомеостазе глюкозы. Она обеспечивает образование более 90 % глюкозы, освобождаемой в печени. При болезни Гирке нарушена одна из важнейших функций печени- поддержание гомеостаза глюкозы крови. Кроме этого нарушен процесс образования глюкозы из аминокислот. Развивающийся обменный дисбаланс приводит к гипогликемии, гиперлактатемии, накоплению гликогена в гепатоцитах. Причиной гипогликемии и увеличения образования лактата являются отсутствие гидролиза Гл-6-Ф и усиление обмена гексозофосфатов в цикле Эмбдена-Мейергофа. Гл-6-Ф накапливается в ткани печени, активирует гликогенсинтетазу и вызывает накопление в печени гликогена, нормального по структуре. В гепатоцитах выявляется большое количество липидов. Нарушение обмена липидов проявляется повышением уровня триглицеридов в крови. Вследствие нарушения энергообеспечения процессов реабсорбции в канальцах почек наблюдаются глюкозурия и аминоацидурия.
Клинические симптомы появляются в 1-й год жизни: маленький рост, "кукольное лицо" (за счет отложения жира в области щек), гипотрофия, гепатомегалия и умеренное увеличение почек. Умственное развитие, как правило, не страдает. Гипогликемия является основным ведущим клиническим симптомом этого типа гликогенозов.
Особенность этого типа гликогенозов: у детей в возрасте 5-7 лет возникают частые носовые кровотечения, геморрагические высыпания, а также гиперурикемия.
Гликогеноз II типа
Открыт H.-G. Hers в 1963 году. Тип наследования аутосомно-рецессивный. Наблюдается дефект фермента кислой a-глюкозидазы.
Это заболевание относится к лизосомальным болезням. При гликогенозе II типа практически во всех органах больного отсутствует активность кислой a-глюкозидазы. Этот фермент расщепляет a-1,4- и a-1,6 связи в молекуле гликогена. При дефекте фермента в лизосомах происходит аномальное накопление гликогена. Эти аномальные лизосомы служат морфологическими маркерами. Болезнь Помпе- единственная лизосомальная болезнь из гликогенозов, остальные гликогенозы связаны с дефектами ферментов, локализованных в цитоплазме.
Заболевание чрезвычайно гетерогенное. Различают инфантильную форму (болезнь Помпе), юношескую и взрослую формы. Каждая из форм гликогеноза II типа неоднородна в связи с существованием различным вариантов дефекта в синтезе или процессинге фермента.
Генерализованная форма заболевания - болезнь Помпе (инфантильная форма гликогеноза II типа, гликогеноз типа IIа). При болезни Помпе с 1-го года жизни наблюдается симптомокомплекс сердечной недостаточности. Поэтому этот тип заболевания часто называют "сердечным гликогенозом". У больного выявляется "шаровидное" сердце, умеренная гипертрофия мышц, увеличение печени, прогрессирующая мышечная слабость. Гипертрофия мышечных волокон сопровождается большим накоплением гликогена. Смерть на 1-ом или 2-ом году от сердечной недостаточности и респираторных заболеваний. При патологоанатомическом исследовании обнаруживают гипертрофию миокарда без органического поражения клапанов.
При гликогенозе II типа наблюдается накопление гликогена не только в печени, скелетных и сердечной мышцах, но и в легких, селезенке, гипофизе, надпочечниках, диафрагме, мозге, в тканях глаза. Структура гликогена при этом не отличается от нормы. Гликоген может находится в лизосомах (аномальные лизосомы) или же свободно лежать в цитоплазме гепатоцитов при разрушении лизосом. Единственным органом, в котором не обнаруживается значительное накопление гликогена, являются почки (в них имеется особый нейтральный фермент, активный в кислых условиях).
При юношеской форме наблюдается дефицит активности a-глюкозидазы в мышцах. Смерть наступает в результате осложнений респираторного характера.
При взрослой форме заболевание напоминает прогрессирующую мышечную дистрофию Дюшена. Клинические симптомы менее выражены, у больных поражаются только скелетные мышцы.
Особенность гликогеноза II типа- отсутствие у больных нарушений обмена. Это связано с тем, что фосфорилазная система не нарушена. Пренатальный диагноз основан на обнаружении вакуолизированного гликогена в амниотической жидкости и исследовании активности фермента в биоптатах хориона.
Гликогеноз III типа
(Болезнь Кори, болезнь Форбса, лимитдекстрино з)
Наследуется по аутосомно- рецессивному типу.
Это заболевание вызывается полным отсутствием или снижением активности амило-1,6- глюкозидазы- фермента, расщепляющего a- 1,6- глюкозидные связи в точках ветвления гликогена.
Дефект данного фермента приводит к образованию аномального по структуре и свойствам гликогена- лимитдекстрина, сходного по структуре с декстрином. Лимитдекстрин имеет укороченные концевые ветви молекул. Аномальный гликоген накапливается в печени и является "инертным" для обмена. При гистохимическом исследовании гепатоцитов обнаруживают их жировую инфильтрацию и накопление полисахаридов в цитоплазме. При световой микроскопии печени выявляют ядерный гликогеноз с небольшим количеством внутриклеточных включений. Необходимо отметить, что амило-1,6 глюкозидаза имеет 2 ферментативные активности: трансферазную и гидролитическую. Такая сложность функций фермента является причиной существования различных форм заболевания (генерализованная и локализованная). Синтез данного фермента в печени и мышцах контролируется различными генами, поэтому при нарушении ферментативной активности лимитдекстрин накапливается только в печени или в мышцах (в саркоплазме, между миофибриллами). Нарушение процесса распада гликогена сопровождается активацией компенсаторных механизмов. Активизируется распад белков, образуются глюкогенные и кетогенные аминокислоты.
Наиболее тяжелые проявления гликогеноза II типа наблюдаются при генерализованной форме.
Клинические проявления гликогенеза III типа: гепатомегалия, задержка физического развития (без нарушения интеллекта), гипогликемия натощак, т.е. клиническая картина у детей часто напоминает болезнь Гирке. Заболевание протекает доброкачественно. Оно наиболее опасно в возрасте 4-5 лет, когда наблюдаются частые приступы гипогликемии, ацетонемия и метаболический ацидоз. У взрослых обнаруживается мышечная слабость и сердечная недостаточность.
Гликогеноз IV типа
(болезнь Андерсена, амилопектиноз)
Клиническая картина заболевания впервые описана Andersen в 1956 году. Заболевание наследуется по аутосомно- рецессивному ти-
пу. При гликогенозе IV типа наблюдается дефект фермента амило-1,4---> 1,6- трансглюкозидазы, участвующего в образовании точек ветвления в молекуле гликогена:
При гликогенозе IV типа в пораженных органах синтезируется аномальный гликоген, подобный амилопектину (компоненту крахмала растительных клеток). Молекула аномального гликогена имеет уменьшенное число точек ветвления и более длинные наружные и внутренние цепи по сравнению с нормой.
Болезнь встречается редко, носит генерализованный характер (чаще поражаются сердечная и скелетная мускулатура, печень). Клинически заболевание проявляется гепатоспленомегалией, асцитом, умственное развитие не страдает. Прогрессирующий портальный фиброз печени приводит к циррозу . Цирроз, возможно, развивается в результате накопления амилоподобного гликогена.
Смерть в детском возрасте от печеночной недостаточности. При патологоанатомическом исследовании обнаруживается увеличение размеров почек, печени, селезенки. Гепатоциты увеличены в размерах и содержат амилопектиноподобный полисахарид.
Болезнь Лафора - гликогеноз мозга (миоклоническая эпилепсия). При этом заболевании обнаруживается накопление в мозге аномального гликогена, напоминающего по свойствам полимер при гликогенозе IV типа. Активность ветвящего фермента при этом заболевании не изменена.
Гликогеноз V типа
(болезнь Мак-Ардля)
Впервые описан B.McArdle в 1951 г. Аутосомно- рецессивный тип наследования. Характеризуется недостаточностью мышечной фосфорилазы в скелетных мышцах. Отсутствие мышечной фосфорилазы не сочетается с нарушением печеночной фосфорилазы (контролируются различными генами). Активность фосфорилазы лейкоцитов, эритроцитов, тромбоцитов при болезни Мак-Ардля не изменена.
При этом заболевании в мышечных волокнах накапливается до 3-4 % нормального по структуре гликогена. Избыточный гликоген откладывается под сарколеммой в цитоплазме. В состоянии покоя энергетические потребности обеспечиваются за счет глюкозы миоцитов. При мышечной работе потребность в энергетическом обеспечении не восполняется за счет ферментативного дефекта, что вызывает боли и судороги при данном типе гликогеноза.
Болезнь Мак-Ардля гетерогенна. Клинические признаки чаще проявляются у взрослых, в детском возрасте симптомы заболевания не выражены. Заболевание протекает в 3 стадии:
1. В детском и юношеском возрасте наблюдаются мышечная слабость,утомляемость, возможна миоглобинурия.
2. В возрасте от 20 до 40 лет мышечные боли становятся более интенсивными, характерно появление судорог после физической нагрузки.
3. После 40 лет возникает прогрессирующая слабость на фоне мышечной дистрофии.
Установлено, что активность фосфорилазы резко снижается при авитаминозе В6 (60% пиридоксина в скелетных мышцах связано с фосфорилазой). Поэтому дефицит фосфорилазы отражается на содержании пиридоксина в организме. Прогноз при гликогенозе V типа благоприятный.
Гликогеноз VI типа
(болезнь Херса)
Впервые описан H.-G.Hers в 1959 году. Аутосомно-рецессивный тип наследования.
При данном заболевании наблюдается дефицит печеночной фосфорилазы. В мышцах активность фосфорилазы не изменяется.
Клиническая картина гликогеноза VI типа характеризуется выраженной гепатомегалией, печень выступает из-под края реберной дуги на 6-9 см (гепатомегалия с возрастом уменьшается). У большинства детей гипогликемия отсутствует.
Характерным признаком заболевания является повышение содержания холестерина, нейтральных жиров и активности трансаминаз в сыворотке крови. Содержание катехоламинов в моче не изменено. Умственное развитие при болезни Херса не страдает. Прогноз при гликогенозе VI типа благоприятный.
БОЛЕЗНИ НАКОПЛЕНИЯ ЛИПИДОВ
Нервная ткань содержит большое количество различных липидов. Цереброзиды, ганглиозиды и сфингомиелины относятся к сфинголипидам, важнейшей функцией которых является обеспечение миелинизации нервных волокон. Сфингомиелины содержатся преимущественно в нервной ткани и состоят из сфингозина, остатков жирных кислот, фосфорной кислоты и холина. Цереброзиды содержат сфингозин, жирные кислоты, углеводы (галактозу или глюкозу). Они входят в состав мозга. Ганглиозиды- сложные липиды, состоят из сфингозина, жирных кислот, гексозы, гексозаминов, нейраминовых кислот. В мозговой ткани находится до 90 % ганглиозидов. Миелинизация- процесс, отражающий морфологическую и функциональную зрелость нервной системы.
Липиды миелина обладают изоляционными свойствами для нормального проведения нервного импульса. Ведущую роль в миелинизации нервных волокон играют сфинголипиды. Первые признаки миелинизации в спинном мозге выявляются на 22-23 неделе внутриутробной жизни. У новорожденного миелинизирована нервная система до оральных отделов ствола головного мозга. Наиболее активно процессы миелинизации протекают в первые два года жизни и постепенно заканчиваются к 16-20 годам. По химическому составу мозговая ткань ребенка 3-х лет идентична таковой у взрослых. Но за период миелинизации масса мозга увеличивается в 2,5 раза.
Демиелинизация наблюдается при токсических и воспалительных поражения нервной системы.
Типы нарушения миелинизации:
1. Недостаточное образование миелина.
2. Изменение структуры миелина.
3. Демиелинизация - повышенный распад миелина.
4. Дисмиелинизация - повышенный распад аномально построенного миелина.
Ферментный блок или снижение активности ферментов может приводить к внутриклеточным липидозам или сопровождаться нарушением проводящих путей мозга.
Таблица 3 Накопление липидов при болезнях накопления липидов
| Заболевание | Накопившийся липид | Орган накопления |
| Болезнь Тея-Сакса | Ганглиозид GМ2 Азиалоганглиозид GА2 |
Мозг |
| Заболевание | Накопившийся липид | Орган накопления |
| Болезнь Сандхоффа | Ганглиозид GМ2 Азиалоганглиозид GА2 Глобазид |
Мозг Печень Печень,селезенка, почки |
Генерализованный GМ1- ганглиозидоз |
Ганглиозид GМ1 Азиалоганглиозид GА1 Ганглиозид GМ1 (мукополисахариды) |
Мозг Висцеральные органы |
| Болезнь Гоше | Глюкоцереброзиды | Печень,селезенка, лимфатические узлы |
| Болезнь Краббе | Галактоцереброзиды | Мозг |
| Метахроматическая лейкодистрофия | Сульфатиды | Мозг, почки |
| Болезнь Фабри | Церамидтригексозид | Почки, кожа |
| Лактозилцерамидоз | Лактозилцерамид | Печень, мозг, почки, кровь |
Болезнь Ниманна- Пика |
Сфингомиелин, холестерин | Селезенка,печень, мозг |
| Болезнь Рефсума | Фитиновая кислота | Кровь, висцеральные органы |
ГАНГЛИОЗИДОЗЫ
GМ 1 - ганглиозидозы:
а) тип I - семейный нейровисцеральный липидоз;
б) тип II - ювенильный системный липидоз (болезнь Дерри).
GМ 2 - ганглиозидозы:
а) болезнь Тея-Сакса;
б) болезнь Сандхоффа;
в) юношеская форма.
Семейный нейровисцеральный липидоз
Аутосомно-рецессивный тип наследования. Основной дефект -недостаточность b-галактозидазы, отщепляющей концевую галактозу от GМ2 -ганглиозида.
У детей выявляется задержка психомоторного развития, мышечная гипотония, слабость сосания и крика. Наблюдаются характерные изменения лица и костей: гипертелоризм, выступающий лоб, гипертрофия десен, кифосколиоз, сгибательные контрактуры суставов. Происходит вакуолизация клеток костного мозга и лимфоцитов. Наблюдается нарастание GМ1 ганглиозида в клетках головного мозга, а также мукополисахаридов и ганглиозидов во всех органах и тканях.
В начале 2-го года жизни ребенка появляются тонико-клонические судороги, затруднения при глотании, в дальнейшем развивается децеребральная ригидность. Смерть наступает от сопутствующих бронхолегочных инфекций. Лечение не разработано.
Болезнь Тея- Сакса
Американский врач W.Tay в 1881 году наблюдал ребенка в возрасте 1 года с выраженной слабостью мышц туловища и конечностей, Sashs назвал синдром амавратической семейной идиотией.
Болезнь Тея-Сакса является GМ2 - ганглиозидом, при котором имеется дефект N-ацетил- b- D- гексозаминидазы А.
Клиническая картина довольно типична: до 4-5 месяцев ребенок развивается нормально. После простудных заболеваний, прививок дети становятся апатичными, вялыми, перестают сидеть и держать голову, узнавать мать. В течение 2-4 мес. развивается тяжелый дефект психики: бессмысленный, нефиксированный взгляд, амимичное лицо, беспричинная улыбка. Постоянным симптомом, выявляющимся с рождения или в первые месяцы, является феномен "гиперакузии"- повышение двигательной реакции на слуховые раздражители. У таких детей отмечается плохой сон, приступы насильственного смеха или плача, нередко заканчивающиеся судорогами, мышечный гипертонус, позднее прорезывание зубов, раннее появление симптома "вишневого пятна или косточки" на глазном дне, который развивается вследствие атрофии ганглиозных клеток сетчатки и просвечивания сосудистой оболочки. Обычно параллельно распаду психических функций нарастают значительные нарушения со стороны двигательной сферы: слабость мышц, обездвиженность, тетрапарез. Наблюдается характерное для болезни Тея-Сакса изменения тонуса мышц: симптом "трупного мешка"- если ребенка взять на руки, голова и конечности свешиваются как плети.
При болезни Тея-Сакса в нейронах (главным образом в ЦНС) содержатся мембранозные цитоплазматические тельца. Наблюдается пролиферация микроглии. В спинном мозге аналогичные изменения наиболее выражены в клетках передних рогов.
Постоянным проявлением болезни Тея-Сакса является гидроцефалия, хотя при люмбальной пункции, давление спиномозговой жидкости может быть нормальным или пониженным, т.е. гидроцефалия носит характер ex vacuo ( происходит заполнение жидкостью пространств, освобождаемых атрофирующимися участками мозга). При исследовании липидов в ткани головного мозга выявляется повышение содержания сфингомиелина и цереброзидов в сером веществе, значительное снижение уровня холестерина, ФЛ и цереброзидов в белом веществе. Это свидетельствует о нарушении процессов миелинизации. Изменение метаболизма миелина определяется накоплением в мозге ганглиозидов (уровень GМ2 ганглиозида увеличивается в 100 раз).
Сфингомиелинозы
Болезнь Ниманна-Пика
Аутосомно-рецессивное наследование.
В 1914 г. А.Niеmann описал поражение нервной системы у девочки из еврейской семьи, сочетавшееся со спленомегалией, и расценил его как частный случай болезни Гоше, т.к. в селезенке обнаружил большое количество клеток, похожих на клетки Гоше. L.Pick выявил гистологические отличия данного заболевания и назвал его липоидно- клеточной спленомегалией. Заболевание чаще встречается среди еврейского населения. Биохимический дефект развивается в результате дефицита активности фермента сфингомиелиназы, катализирующего гидролиз фосфохолина из сфингомиелина. Известно по А. Crocker (1961 г) 4 типа болезни Ниманна-Пика (А, В, С, Д). В 1964 -1978 гг выделили еще 2 типа: Е и F. Дефицит активности сфингомиелиназы выявляется при типах А,В,F, но не обнаруживается при С,Д и Е. Чаще встречается классическая (инфантильная) форма болезни (тип А). Проявляется заболевание в возрасте 5-6 месяцев с изменения поведения: вялость сменяется периодами психомоторного раздражения. Возможны периоды кратковременной асфиксии, развиваются гипертермические кризы, пирамидные симптомы, атрофия зрительных нервов, в 1/3 случаев обнаруживается симптом "вишневой косточки", гепатоспленомегалия. Течение этой формы болезни неуклоннопрогрессирующее, развивается вегетативное состояние, смерть наступает в возрасте 4-х лет. Морфологически отмечается увеличение массы мозга, уплотнение его вещества, стирание границ между серым и белым веществом. Гистологическая картина: клетки КБП и мозжечка баллоно-образно вздуты, содержат большое количество сфиногомиелина. Аналогичные изменения в клетках спинного мозга. В легких, печени, селезенке- клетки Ниманна-Пика с одним или несколькими ядрами, пенистой цитоплазмой.
Тип В ювенильный- проявляется висцеральной патологией в виде гепатоспленомегалии. Во внутренних органах увеличено количество сфингомиелина и холестерина.
Тип С- позднее начало, хроническое течение, поражаются внутренние органы и нервная система.
Тип Д- висцеральные проявления.
Тип Е - описан у взрослых с хроническим течением заболевания. Тип F - спленомегалия и неврологические нарушения в детском возрасте.
ГЛЮКОЦЕРЕБРОЗИДОЗЫ
Болезнь Гоше
Впервые была описана Gaucher в 1882 г., который кроме гепатоспленомегалии, пигментации кожи, обратил внимание на наличие больших клеток в селезенке и считал их доказательством первичного неоплазменного процесса в этом органе. Лишь спустя 40 лет, в 1924 г. Epstein обнаружил патологию обмена липидов при болезни Гоше, а в 1934 г. Aghion установил, что накапливающийся цереброзид является глюкоцереброзидом.
Метаболический дефект при болезни Гоше развивается вследствие дефицита фермента глюкоцереброзидазы, катализирующего отщепление глюкозы от глюкоцереброзидов. Этот фермент находится в различных органах и тканях.
Три клинических типа болезни Гоше:
1. Взрослый хронический висцеральный (аутосомно-доминантный тип наследования). Начало заболевания в детстве. Характеризуется медленным течением, анемией, тромбоцитопенией, кровоизлияниями. Смерть- в 20-50 лет.
2. Раннедетский острый нейровисцеральный (этнически распространен в еврейских семьях, аутосомно- рецессивный тип наследования). Начало на 1-ом году жизни, быстропрогрессирующее поражение мозга, гепатоспленомегалия. Дети умирают до 2-х летнего возраста от интеркуррентных заболеваний.
3. Юношеский подострый нейровисцеральный (начало в юношеском возрасте). Задержка умственного развития, гепатоспленомегалия, поражение пирамидной и экстрапирамидной системмозга.
Патологоанатомические изменения при болезни Гоше характеризуются поражением печени, селезенки, костного мозга, лимфатических узлов. В них выявляются клетки Гоше. Клетки Гоше- гистиоциты, содержащие глюкоцереброзид. Они имеют одно или несколько эксцентричных ядер, голубая цитоплазма похожа на смятый шелк. В мозгедиффузные изменения в пирамидных и ганглиозных слоях коры с участками некроза. Содержание цереброзидов в мозге увеличено, содержание липидов миелина - снижено.
До недавнего времени радикальными методами лечения были трансплантация костного мозга и генотерапия. Применение фермента альглюцеразы (цередаза) или рекомбинантной имилглюцеразы (церезим) - аналогов естественной глюкоцереброзидазы позволило улучшить качество жизни больных, остановить поражение висцеральных органов, смягчить гематологические проявления болезни.
Болезнь Фабри
Наследуется сцепленно с Х-хромосомой. Причиной заболевания является дефект фермента a-галактозидазы А.
Ранее болезнь Фабри называлась диффузной ангиокератомой, так как ангиокератомы, расположенные по типу "купального костюма", считали патогномичным признаком.
Образующиеся в результате дефекта фермента продукты (тригексозил- и дигалактозилцерамид) накапливаются в почечных канальцах, в сердечной мышце, в скелетных мышцах, в нейронах головного мозга, в эндотелии всех сосудов. В пораженных тканях обнаруживают пенистые клетки и суданофильные гранулы.
Накопление гликолипида вызывает характерные симптомы: быструю утомляемость, снижение остроты зрения, повышение артериального давления.У детей возникают приступы лихорадки, боли в руках и ногах, протеинурия, ангиокератомы. Продолжающееся накопление липидов приводит к почечной и сердечной недостаточности в возрасте 30-40 лет.
Диагноз основан на снижении активности a-галактозидазы в
плазме, моче, лейкоцитах, культуре клеток кожи.
ЛЕЙКОДИСТРОФИИ
Лейкодистрофии - заболевания, возникающие в результате генетически детерминированных нарушений обменных процессов миелина с его последующим распадом и характеризующиеся диффузной дегенерацией белого вещества головного и спинного мозга.
Болезнь Краббе
Синонимы: галактозилцерамидный липоидоз, глобоидно-клеточная лейкодистрофия. В 1916 г. Krabbe впервые описал данное заболевание.
У больных наблюдается дефицит галактозилцерамид b-галактозидазы в мозге, лейкоцитах крови, сыворотке, селезенке, печени. В норме галактозилцерамид- b-галактозидаза катализирует метаболизм галактозилцерамида, отщепляя галактозу и образуя церамид, от которого ферментом церамидазой отщепляется жирная кислота с образованием сфингозина. Генетический блок активности фермента обусловливает накопление больших количеств галактоцереброзида, что приводит к нарушению миелинизации нервных волокон ("дисмиелинизации") и к демиелинизации. Лейкодистрофия Краббе является гетерогенной группой заболеваний, сходных клинически и патоморфологически, однако, разнородных биохимически. Так, ряд авторов считает, что при этой болезни увеличивается содержание психозина, который локально достигает токсического уровня и задерживает миелинизацию. Это приводит к гибели олигодендроглиальной клетки.
Клинически до 3-6 месяцев дети развиваются нормально, затем наблюдается остановка и ухудшение психического и физического развития. Возникают приступы крика, развивается слабость в конечностях. Затем развивается состояние опистотонуса с клоническими судорогами в конечностях. Могут наблюдаться гипертермические приступы без инфекции, атрофия зрительных нервов. Заключительный период характеризуется развитием умственной отсталости, децеребрационной ригидностью, кахексией, слепотой. В типичных случаях через 5-8 месяцев от начала заболевания больные умирают.
Патоморфологически наблюдается уменьшение размеров и массы мозга. Гистологически: выраженная демиелинизация, уменьшение количества олигодендроглиальных клеток. В демиелинизированном белом веществе- глобоидные клетки, располагающиеся изолированно или группами вокруг сосудов. Эти клетки имеют сферическую форму, множественные ядра, сдвинутые к периферии клетки. Рядом с глобоидными присутствуют эпителиоидные клетки. Гистохимически оба вида клеток дают идентичные реакции на красители за счет наличия галактоцерамида.
Таблица 4 Биохимическая диагностика болезней накопления липидов
| Заболевание | Параметр | Материал исследования |
| Болезнь Тея-Сакса | Гексозаминидаза А Ганглиозид GМ2 Азиалоганглиозид GА2 |
Сыворотка, лейкоциты, фибробласты кожи, материал биопсии органов, моча. Материал биопсии мозга. |
| Болезнь Сандхоффа | Гексозаминидаза А и В Глобазид |
Материал биопсии органов, сыворотка, фибробласты. Материал биопсии мозга,осадок мочи. |
Генерализованный GМ1- ганглиозидоз |
b- Галактозидаза Ганглиозид GМ1 |
Лейкоциты, материал биопсии, фибробласты кожи Материал биопсии |
| Болезнь Гоше | b- Глюкозидаза (глюкоцереброзидаза) Глюкоцереброзиды |
Материал биопсии органов, лейкоциты, фибробласты кожи. Материал биопсии органов. |
| Болезнь Краббе | b- Галактозидаза | Сыворотка, лейкоциты, фибробласты кожи. |
| Метахроматическая лейкодистрофия | Арилсульфатаза Сульфатиды |
Лейкоциты, фибробласты кожи, моча. Моча |
| Болезнь Фабри | a-Галактозидаза (церамидтригексозидаза) Церамидтригексозид |
Лейкоциты, биопсия органов, фибробласты кожи, моча. Материалы биопсии почек, моча. |
| Лактозилцерамидоз | b- Галактозидаза Лактозилцерамид |
Фибробласты кожи, материал биопсии печени. Материал биопсии органов, плазма, эритроциты, осадок мочи. |
Болезнь Ниманна- Пика |
Сфингомиелиназа Сфингомиелин, холестерин. |
Лейкоциты, материал биопсии органов, фибробласты кожи (инфантильная и ювенильная формы) Материал биопсии |
Гиперлипопротеидемии (ГЛП)
Наследственные заболевания нарушений липидного обмена представлены большим количеством нозологических форм, которые обусловливают развитие атеросклероза и других форм сердечно-сосудистой патологии.
Впервые гиперлипидемии описаны еще в конце ХIХ в. Их часто связывали с ксантоматозом и сердечно- сосудистыми расстройствами. Первичные нарушения липидного обмена, семейные гиперлипопротеидемии, являются группой наследственных болезней, обусловленных различными мутациями, характеризуются специфическим подъемом уровня Холестерина (ХС) и триглицеридов (ТГ) в плазме крови за счет измененного содержания основных классов липопротеидов (ЛП) плазмы крови (ЛПОНП,ЛПНП, ЛПВП и др.). Наследственные формы ГЛП делятся на моногенные и полигенные (табл. 5).
Таблица 5. Классификация семейных гиперлипидемий
| ГЛП | Фенотип | Концентрация липидов в сыворотке крови | Концентрация ЛП в сыворотке крови | Метаболи-ческий дефект | |
| ХС | ТГ | ||||
| Семейная Недостаточность ЛПЛ, гиперхиломикронемия. | I | Нормальная или незначит. повышенная | Повышенная | 1)ХМ 2)ЛПОНП, ЛПНП, ЛПВП- нормальная или незначительно повышенная |
Дефект Апо-С или недостаточность ЛПЛ |
| Семейная Гиперхолестеринемия | IIа | Повышенная | Нормальная | 1)ЛПНП-повышенная 2)ЛПОНП- нормальная |
Дефект или отсутствие апо-В Отсутствие клеточных рецеп торов апо-В или апо-С. |
| Семейная комбинированная ГЛП | IIб в сочетании с IIа и (или) IV | Повышенная | Повышенная | ЛПНП, ЛПОНП повышенНая | Дефект интернализации. |
| Дис-b-липо-про теидемия | III | Повышенная | Повышенная | Обнаружиются "патологические"ЛПОНП | Нарушение структуры апо-Е |
| Семейная гипертри глицеридемия | IV | Нормальная или незначит. повышенная | Повышенная | 1)ЛПОНП - повышенная 2)ХМ отсут 3)ЛПНП – нормальная |
Молекулярный дефект неизвестен. |
| Семейная ГЛП V ти па (гипертриглицеридемия с гиперхиломикронемией | V | Повышенная | Повышенная | 1)ЛПОНП-повышенная 2)ХМ присутствует |
Дефект или отсутсвие апо-С, дефект или недостаточность ЛПЛ |
В 1967 г Fredrickson с соавт. предложили классификацию гиперлипопротеидемий. Из 5 выделенных ими типов ГЛП только 3 относились к моногенным заболеваниям, а 2 являлись полигенными (I и V типы). Впоследствии эта классификация была модифицирована ВОЗ и нашла широкое применение.
Гиперлипопротеидемия I типа
Считается редким заболеванием (известно около 100 случаев). Аутосомно- рецессивный тип наследования. ГЛП I типа развивается в результате низкой активности липопротеидлипазы (мутация 207 остатка в пятом экзоне гена липопротеидлипазы) или при недостатке аполипоротеина С II (активатора липопротеидлипазы).
Недостаточная активность липопротеидлипазы характеризуется сниженным метаболизмом хиломикронов. Нередко заболевание диагностируется случайно, при биохимическом исследовании крови, когда наблюдается выраженная гиперхиломикронемия (10-40 г/л). Гиперхиломикронемия связана с неспособностью хиломикронов гидролизоваться.
Клинический симптомокомплекс: панкреатит, абдоминальные колики, гепатоспленомегалия. Возможны лихорадка, лейкоцитоз, ретинальная липемия и эруптивный ксантоматоз. Атеросклероз у таких больных обычно не развивается.
Боли в области живота чаще всего первая жалоба у таких больных. Механизм развития этих болей и панкреатита при гиперхиломикронемии остается загадкой (одной из гипотез является закупорка микрососудов поджелудочной железы агрегатами ХМ, развивается ишемия, что приводит к локальному высвобождению панкреатических ферментов). При морфологическом исследовании печени и селезенки выявляется большое количество "пенистых" клеток, содержащих липиды, наблюдается вакуолизация паренхимы печени и купферовских клеток.
Ксанотоматозные высыпания на эритематозном фоне свидетельствуют о резко выраженной гиперхиломикронемии (>40 г/л). Они не вызывают зуда, локализуются на туловище, разгибательных поверхностях предплечий, ягодицах, бедрах. После уменьшения уровня ТГ ксантомы через 1-3 месяца исчезают. Другим признаком заболевания является липемия сетчатки (артерии и вены при исследовании глазного дна имеют цвет семги).
В сыворотке крови повышенный уровень ТГ (концентрация возрастает в 8-10 раз) сопровождается значительно меньшим повышением ХС (соотношение обычно превышает 10:1). Высокая насыщенность ХМ придает сыворотке молочный или кремовый цвет.
Диагноз основан на определении постгепариновой липолитической активности.
Гиперлипопротеидемия II типа (Семейная гиперхолестеринемия)
Синоним: семейная ксантоматозная гиперхолестеринемия. Относится к одной из наиболее распространенных форм ГЛП.Наследуется аутосомно- доминантно.
Ведущим звеном патогенеза гиперхолестеринемии, высокого содержания в плазме крови атерогенных липопротеидов низкой плотности является или полное отсутствие рецепторов к липопротеидам на наружной клеточной поверхности или нарушение их строения и функции вследстивие мутации аллелей генов RbO, Rb- и Rito. В результате наследуемой недостаточности рецепторов к ЛПНП возникает гиперхолестеринемия, которая по механизму положительной обратной связи повышает синтез атерогенных липопротеидов в печени.
Так как у гетерозигот отмечается дефицит этих рецепторов, а у гомозигот они практически отсутствуют, точная характеристика степени недостаточности рецепторов желательна у гомозигот. Гетерозиготная форма редко приводит к смерти до детородного возраста, передается из поколения в поколение, что и объясняет длительную родословную данного заболевания. Встречается у лиц обоего пола, в любом возрасте и характеризуется резко выраженным кожным и сухожильным ксантоматозом. Инфильтрация кожи и сухожилий ЛПНП с высоким содержанием ХС и его эфиров вызывает легкую воспалительную реакцию тканей и сопровождается накоплением коллагена. Ксантомы на веках называются ксантелазмами. Ксантомы локализуются в области ахилловых сухожилий на разгибательных поверхностях. У подростков может быть пяточный тендинит.
В большинстве случаев отмечается длительный латентный период, когда кроме гиперхолестеринемии заболевание клинически ничем не проявляется.
При этом типе заболевания существует тесная взаимосвязь обменных нарушений с развитием ранних форм атеросклероза. Гомозиготы обычно умирают от ИБС, не прожив и 20 лет. У гетерозигот ИБС развивается в возрасте до 40 лет. Причем женщины обычно утрачивают свойственные им в период сохранения менструальной функции преимущества перед мужчинами в отношении ИБС. Кроме этого, у таких больных наблюдаются и другие проявления: инсульт, поражение периферических сосудов, ксантоматозные поражения аортального и митрального клапанов. Эта форма ГЛП связана с ожирением.
Выделяют 2 типа ГЛП II типа: IIа и IIв. При а-типе в крови увеличена концентрация холестерина при нормальных содержаниях ЛПОНП и ТГ. При в-типе концентрация в крови ЛПОНП и ТГ повышена.
Гиперлипопротеидемия III типа
(семейная дисбеталипопротеидемия)
Называется широкой, плавающей b- или дис-b-липопротеидемией. Относится к редким формам заболевания. Аутосомно-доминантный тип наследования. Эта форма была впервые описана Gofman at al. в 1954 г. Названа бугорчатой ксантомой и характеризуется присутствием в сыворотке крови больных аномальных ЛП с чрезвычайно низкой плотностью,("флотирующих" b липопротеидов). Полагают, что первичный дефект связан с нарушением структуры апо-Е, что приводит к снижению связывания апо-Е ЛП с рецепторами печени.
Средний возраст клинических манифестаций болезни равен 40 годам. Чаще встречается у мужчин. Клиническая картина ГЛП III типа характеризуется кожными и сосудистыми нарушениями. Бугорчатосыпеобразный ксантоматоз представляет собой узелковые образования, сливающиеся в более крупные разрастания. Однако, в отличие от других типов , при этой форме заболевания речь идет о планарных ксантомах, располагающихся на подошвах, тыльных поверхностях кистей, кончиках пальцев, в ладонных складках (хanthoma striata palmaris). Эти проявления обнаруживаются уже в раннем детском возрасте.
Сердечно-сосудистые нарушения проявляются в виде ИБС, развиваются атеросклероз коронарных артерий, ожирение, сахарный диабет, жировая дистрофия печени, гиперурикемия. При морфологическом исследовании обнаруживаются отложения большого количества суданофильных пенистых клеток под эндокардом.
Плазма крови больных мутная, наблюдается появление пре-b- ЛП с повышенным содержанием ХС, выявляется увелечение содержания ХС и ТГ. Это единственный вариант ГЛП, чувствительный к ограничениям в диете углеводов.
Гиперлипопротеидемия IV типа
(гиперпре-b-липопротеидемия)
Составляет 17-37 % от общего числа ГЛП.
Тип наследования аутосомно-доминантный с пенетрантностью в первые два десятилетия жизни.
Специфический метаболический дефект до сих пор не ясен. Предполагается наличие генетического дефекта.
Клинические проявления ГЛП IV типа широко варьируют и нерезко выражены. Ксантомы наблюдаются редко и могут спонтанно исчезать при снижении уровня триглицеридов. Чаще беспокоят боли в животе после приема жирной пищи. В 1/4 случаев могут быть гепатомегалия, общее ожирение, сахарный диабет, развитие перемежающейся хромоты. Большинство больных не предъявляет жалоб, и патология обнаруживается при случайном лабораторном обследовании. У больных старше 45 лет наблюдаются клинические проявления атеросклероза, развивается ИБС.
Биохимически отмечается нормальный уровень ХС в плазме крови, а концентрация ТГ в сыворотке крови повышается до 10-15 г/л.
Для дифференциальной диагностики от II и III типов ГЛП делают хроматограмму, на которой обнаруживается нормальная или ослабленная по интенсивности полоса b-ЛП.
Гиперлипопротеидемия V типа
(гиперпре-b-липопротеидемия и гиперхиломикронемия)
Встречается редко, среди всех типов ГЛП составляет 6-7%.
В основе заболевания лежит недостаток липопротеидлипазы.
Патогенез заболевания связывают с замедлением метаболизма ХМ. Клинические проявления сходны с ГЛП I типа: абдоминальные боли, ксантомы, гепатоспленомегалия, панкреатит, ожирение, сахарный диабет. Характерно снижение толерантности к жирам и низкая постгепариновая липолитическая активность.
У больных наблюдается гиперпре-b- ЛП-емия, в сыворотке крови повышено содержание холестерина и триглицеридов.
Синдром Лоуренса-Муна-Барде-идля (ЛМББ)
Аутосомно- рецессивный тип наследования.
Описано около 400 больных с синдромом ЛМББ, частота заболевания 1:160 000. При этом заболевании часты кровные браки.Большинство исследователей относит синдром ЛМББ к наследственным заболеваниям. Некоторые авторы рассматривают синдром ЛМББ как результат нарушенного внутриутробного развития с преимущественным поражением гипоталамо-гипофизарной системы.
При этом синдроме наблюдается клинический полиморфизм. Особенно высока диагностическая ценность таких кардинальных симптомов, как пигментный ретинит, ожирение, умственная отсталость, полидактилия, синдактилия и гипогенитализм. При пигментном ретините отмечается скопление пигмента в виде "костных телец" по периферии, ночная слепота. Ожирение может быть всех 4-х степеней (причем увеличение массы тела обнаруживается уже на 1-ом году жизни). С полным синдромом ЛМББ коррелирует тяжелая умственная отсталость. Если форма абортивная, то интеллект соответствует норме (табл. 6).
Таблица 6 Особенности интеллектуального дефекта при синдроме ЛМББ в зависимости от клинического варианта болезни
(по Ю.И. Барашневу, Ю.Е. Вельтищеву)
| Клинический вариант болезни | Результаты психологического обследования |
| I (наличие 5 кардинальных признаков) | Грубый интеллектуальный дефект в сочетании с выраженными эмоционально-волевыми нарушениями. Недоразвитие познавательной деятельности и грубые нарушения зрительного анализатора. Вялость, заторможенность, инертность. |
| II (без ожирения) | Интеллектуальное недоразвитие, пограничная умственная отсталость. Преобладание в эмоциональной сфере утомляемости и быстрой истощаемости нервных процессов, относительная сохранность способности к обучению. |
| III (без полидактилии и гипогенитализма) | Умственное развитие соответствует норме при наличии слабой переключаемости мыслительных процессов, тенденция к "соскальзыванию" на более доступные задания и пр. |
Гипогенитализм встречается редко. У женщин наблюдается олиго-и аменорея, гипоплазия яичников, дистрофические изменения в них. У мужчин развивается гинекомастия, крипторхизм, тестикулярная гипоплазия, уменьшение libido sexsualis, аплазия яичек. Но женщины и мужчины с данным синдромом способны иметь детей. Синдром ЛМББ относится к заболеваниям с яркой клинической картиной, однако диагноз заболевания ставится крайне редко, а большинство больных наблюдаются у врачей по поводу самой разной патологии. Если это ожирение- у эндокринолога, нарушение зрения- у офтальмологи, умственная отсталость - у психиатора. При рождении ребенок не имеет какой- либо яркой специфической клинической картины. Единственным симптомом заболевания иногда может быть полидактилия. Лечение синдрома симптоматическое.
Семейная недостаточность лецитинхолестеринацилтрансферазы (ЛХАТ)
Встречается достаточно редко, в основном, в Европе. Аутосомно-рецессивный тип наследования.
При этом заболевании нарушен синтез или секреция ЛХАТ печенью. Концентрация свободного холестерина и фосфатидилхолина в плазме превышает норму, уровень триглициридов повышен, снижено содержание эфиров ХС и лизолецитина.
Клинически дефицит ЛХАТ проявляется в раннем детском возрасте только у гомозигот: анемия, протеинурия, помутнение роговицы. Поражение роговицы в виде множества серых точек по периферии напоминает старческую дугу. В костном мозге и селезенке обнаруживаются голубые гистиоциты. Наблюдается преждевременный атеросклероз и прогрессирующая патология почек. Диагностика основана на определении ЛХАТ в плазме. Специфические методы лечения отсутствуют. Назначается диетотерапия с ограничением животных жиров. Проводится гемодиализ, трансплантация почек и роговицы.
Танжерская бол е знь
Название от о.Танжер (шт. Вирджиния), где была впервые обнаружена.
Редкое нарушение, характеризующееся выраженной недостаточностью или отсутствием полноценных ЛПВП в плазме и накоплением эфиров ХС в тканях. Точный молекулярный дефект не установлен. В качестве механизма триглициридемии предполагается отсутствие нормального источника пептидов в ЛПВП.
Клиника: необычные оранжевые или желтые крупные, дольчатые миндалины. Выявляются отложения липидов в печени, селезенке, лимфоузлах. Эфиры ХС откладываются в слизистой прямой кишки, инфильтрируют роговицу. Наблюдаются неврологические симптомы: парастезии, диплопия, слабость, чрезмерная потливость. Повышенный риск развития атеросклероза не отмечается.
Таким образом, при большинстве наследственных заболеваний, несмотря на различный уровень метаболических блоков и качественно иные метаболические расстройства, ведущим симптомом в клинической картине является задержка психомоторного развития и последующая умственная отсталость. Существует определенная точка зрения, что все эти заболевания фенотипически сходны и без специальных лабораторных исследований врач не может их разграничить. Если исходить из характера поражения ЦНС, то это так. Однако, для каждого заболевания свойственно сочетание нарушений нервной системы и других органов и систем,которое нередко создает достаточно специфическую для каждой нозологической формы картину болезни.
Необходимо отметить, что первые описания новых, ранее неизвестных наследственных болезней обмена веществ, были основаны на выявлении и обследовании больных с грубыми нарушениями психомоторного развития. Клинически было невозможно выделить, например, среди больных с катарактой- больных галактоземией, патологией опорно-двигательного аппарата- больных гомоцистинурией. Истинный характер заболеваний устанавливается при использовании биохимических методов диагностики. Например, в настоящее время расшифрован биохимический дефект синдрома Целвегера, который ранее был отнесен к врожденным порокам развития (ВПР). Это открытие стимулирует к поискам первичного биохимического дефекта при моногенно наследующихся ВПР.
Ранняя диагностика нарушений обмена веществ основана на массовом и селективном биохимическом скрининге. В настоящее время общепринято, что в европеоидных популяциях целесообразно проводить массовый скрининг на фенилкетонурию, гипотиреоз, врожденную гиперплазию надпочечников и др. Селективный биохимический скрининг проводят среди детей (и взрослых) при подозрении на наследственные дефекты обмена веществ.
Признаки, позволяющие заподозрить наследственный дефект обмена веществ (по Е.Т. Лильину, Е.А. Богомазову, П.П. Гофман-Кадашникову)
1. Задержка психомоторного развития у детей раннего возраста (умственная отсталость у детей старшего возраста).
2. Неврологические нарушения (судороги, снижение или повышение мышечного тонуса, спастические парезы).
3. Диспепсические явления, непереносимость отдельных продуктов и лекарств, нарушение вскармливания.
4. Нарушение физического развития детей (задержка в прибавке массы тела, неправильный рост, деформация костей туловища и конечностей).
5. Другие частные проявления (катаракта, нарушения слуха, зрения, специфический цвет и запах мочи, кожные проявления).
ЛИТЕРАТУРА
1. Барашнев Ю.И., Вельтищев Ю.И. Наследственные болезни обмена веществ у детей.- Л.: Медицина.- 1978. - 320 с.
2. Калинина Л.В., Гусев Е.И.. Наследственные болезни метаболизма и факоматозы с поражением нервной системы.- М.: Медицина.-1981. -248 с.
3. Лейтес С.М., Лаптева Н.Н. Очерки по патофизиологии обмена веществ и эндокринной системы.- М.Медицина.- 1967. - 425 с.
4. Лильин Е.Т., Богомазов Е.А., Гофман П.Б.-Кадошников. Генетика для врачей. - М.,Медицина. - 1990.
5. Педиатрия. Руководство. Под ред. Р.Е.Бермана, В.К.Вогана. Болезни плода и новорожденного, врожденные нарушения обмена веществ. Пер. с анг.- М.: Медицина.- 1991. - 528 с.
6. Розенфельд Е.П., Попова И.А. Врожденные нарушения обмена гликогена.- М.: Медицина.- 1989. - 240 с.
7. Дж. Теппермен, Х.Теппермен. Физиология обмена веществ и эндокринной системы. Пер. с анг.- М.: Мир.- 1989.
Похожие рефераты:
Лекции - Патофизиология (патофизиология печени)
Литература - Патофизиология (заболевания печени)
Акушерство. Методические рекомендации кафедры
Общая нозология. Типовые патологические процессы
Лечение эндокринных заболеваний
Роль наследственности и среды в наследственной патологии человека
Тесты по патологической анатомии
Ферменты клинической диагностики
Основные понятия анатомии и физиологии человека