| Скачать .docx |
Курсовая работа: Специфика прогнозирования энтальпии образования ароматических органических соединений
Основные методы прогнозирования энтальпий образования органических соединений относятся к ![]() , то есть характеризуют свойство вещества, находящегося в состоянии идеального газа при давлении 1 атм. и температуре 298,15 К, которую часто называют стандартной. Среди методов массовых расчетов
, то есть характеризуют свойство вещества, находящегося в состоянии идеального газа при давлении 1 атм. и температуре 298,15 К, которую часто называют стандартной. Среди методов массовых расчетов ![]() особого внимания заслуживают методы молекулярной механики и аддитивные методы (лат. additio - прибавление). Большинство полуэмпирических методов не обеспечивает требуемого качества прогноза. Неэмпирические методы не дают прямого выхода на энтальпии образования веществ и к тому же до сих пор являются малодоступными для расчета свойств органических веществ со сложным строением молекул.
особого внимания заслуживают методы молекулярной механики и аддитивные методы (лат. additio - прибавление). Большинство полуэмпирических методов не обеспечивает требуемого качества прогноза. Неэмпирические методы не дают прямого выхода на энтальпии образования веществ и к тому же до сих пор являются малодоступными для расчета свойств органических веществ со сложным строением молекул.
В группе методов молекулярной механики нами накоплен значительный опыт по использованию метода ММХ (на базе силового поля Эллинджера). Метод хорошо зарекомендовал себя в приложении к ![]() алканов. Однако уже для ароматических соединений его целесообразно использовать не для расчета энтальпий образования, а для оценки эффектов взаимодействия заместителей в молекуле, т.е. для разностей энтальпий образования изомеров. Для большинства галогенорганических, кислород-, азот- и серосодержащих соединений метод дает смещенные оценки
алканов. Однако уже для ароматических соединений его целесообразно использовать не для расчета энтальпий образования, а для оценки эффектов взаимодействия заместителей в молекуле, т.е. для разностей энтальпий образования изомеров. Для большинства галогенорганических, кислород-, азот- и серосодержащих соединений метод дает смещенные оценки ![]() . Тем не менее, этот метод следует применять во всех случаях для экспрессной оценки свойства. Как и любой другой метод, он требует подкрепления результатов сведениями, полученными другим методом прогнозирования. Метод молекулярной механики имеет прекрасный интерфейс и исключительно результативен как источник информации о строении молекул и их геометрических параметрах.
. Тем не менее, этот метод следует применять во всех случаях для экспрессной оценки свойства. Как и любой другой метод, он требует подкрепления результатов сведениями, полученными другим методом прогнозирования. Метод молекулярной механики имеет прекрасный интерфейс и исключительно результативен как источник информации о строении молекул и их геометрических параметрах.
При отсутствии справочных данных для прогнозирования ![]() в настоящее время широко используются различные аддитивные методы. С момента создания основных аддитивных методов прогнозирования свойств органических веществ, находящихся в состоянии идеального газа, прошел значительный период времени, однако они сохраняют свою значимость, несмотря на становящиеся все более доступными методы молекулярного моделирования. Эти методы эффективны в тех случаях, когда свойство изменяется линейно при изменении количества однотипных фрагментов в молекуле. Строго аддитивной является, например, молекулярная масса вещества. Для энтальпий образования органических соединений аддитивный подход является во многих случаях лишь некоторым приближением в расчете. Дело в том, что даже в гомологической группе
в настоящее время широко используются различные аддитивные методы. С момента создания основных аддитивных методов прогнозирования свойств органических веществ, находящихся в состоянии идеального газа, прошел значительный период времени, однако они сохраняют свою значимость, несмотря на становящиеся все более доступными методы молекулярного моделирования. Эти методы эффективны в тех случаях, когда свойство изменяется линейно при изменении количества однотипных фрагментов в молекуле. Строго аддитивной является, например, молекулярная масса вещества. Для энтальпий образования органических соединений аддитивный подход является во многих случаях лишь некоторым приближением в расчете. Дело в том, что даже в гомологической группе ![]() изменяется нелинейно с изменением числа углеродных атомов в молекуле (рис. 1.1).
изменяется нелинейно с изменением числа углеродных атомов в молекуле (рис. 1.1).
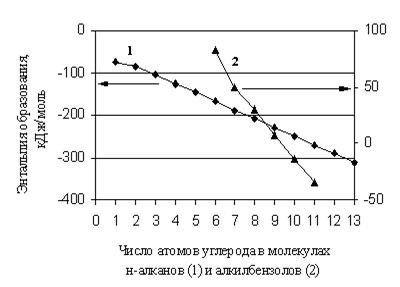
Рис. 1.1. Зависимость энтальпии образования н-алканов и алкил бензолов от числа атомов углерода в их молекулах
Таким образом, гомологическая разность не является величиной постоянной, особенно для первых членов гомологических групп. Точно так же при увеличении количества заместителей одного вида в молекулах органических веществ очень часто приходится говорить об отклонении от аддитивности в ![]() . Однако при введении поправок на неаддитивность методы данной группы работают вполне удовлетворительно, если степень и глубина их детализации достаточны и отвечают точности современного эксперимента.
. Однако при введении поправок на неаддитивность методы данной группы работают вполне удовлетворительно, если степень и глубина их детализации достаточны и отвечают точности современного эксперимента.
В зависимости от принятой идеологии в качестве носителя структурной и количественной информации в аддитивных методах могут выступать составляющие молекулу атомы, группы атомов или связи. Большинство методов прогнозирования построено таким образом, что по мере расширения базы данных по энтальпиям образования относительно легко могут быть уточнены значения парциальных вкладов или введены новые поправки.
Общий подход к прогнозированию энтальпий образования веществ предполагает вычисление ![]() с последующим, при необходимости, переходом к идеально-газовым энтальпиям образования при других температурах или к
с последующим, при необходимости, переходом к идеально-газовым энтальпиям образования при других температурах или к ![]() , т.е. к свойству вещества в реальном состоянии.
, т.е. к свойству вещества в реальном состоянии.
Следует признать, что из всего многообразия аддитивных схем для прогнозирования энтальпий образования органических веществ метод Бенсона в течение продолжительного периода применяется наиболее широко. Объясняется это, вероятно, тем, что этим методом охвачен наиболее широкий круг соединений. Для оперативной оценки ![]() абсолютного большинства соединений без привлечения каких-либо технических средств метод, пожалуй, не имеет себе равных. Совершенно очевидно, что ценой его универсальности является точность прогноза. Поэтому при использовании метода необходимо знать о неизбежных его ограничениях. На основные из них, являющиеся результатом нашей широкой апробации метода, мы постараемся обратить внимание потенциальных пользователей.
абсолютного большинства соединений без привлечения каких-либо технических средств метод, пожалуй, не имеет себе равных. Совершенно очевидно, что ценой его универсальности является точность прогноза. Поэтому при использовании метода необходимо знать о неизбежных его ограничениях. На основные из них, являющиеся результатом нашей широкой апробации метода, мы постараемся обратить внимание потенциальных пользователей.
Метод Бенсона принято называть групповым, хотя в качестве структурной единицы в нем избран атом с его первым окружением. Метод разработан автором для расчета следующих идеально-газовых свойств веществ: теплоемкости ![]() при температурах, кратных 100 градусам, энтальпии образования
при температурах, кратных 100 градусам, энтальпии образования![]() и энтропии
и энтропии ![]() .
.
Алкилбензолы и их функциональные производные
Объем экспериментальных сведений для ![]() ароматических углеводородов существенно меньше, чем для алканов, и строение молекул соединений, для которых имеются надежные калориметрические данные, не отличается большим разнообразием. Это не позволяет в настоящее время выработать подходы к прогнозированию их энтальпий образования, опираясь только на калориметрические данные. Для этой цели нами использована вся совокупность фактического материала и возможности неэмпирических методов расчета энергии и геометрии молекул, а также метода молекулярной механики с силовым полем Эллинджера.
ароматических углеводородов существенно меньше, чем для алканов, и строение молекул соединений, для которых имеются надежные калориметрические данные, не отличается большим разнообразием. Это не позволяет в настоящее время выработать подходы к прогнозированию их энтальпий образования, опираясь только на калориметрические данные. Для этой цели нами использована вся совокупность фактического материала и возможности неэмпирических методов расчета энергии и геометрии молекул, а также метода молекулярной механики с силовым полем Эллинджера.
В результате можно с уверенностью говорить о том, что при использовании любого аддитивного метода для ![]() алкилароматических углеводородов необходимо вводить поправки, природа которых в основном имеет стерическое происхождение. Величина этих поправок никоим образом не является постоянной, как это принято, например, в методе Бенсона, а зависит от эффективных размеров взаимодействующих групп, от количества рядом расположенных заместителей и от их взаимной ориентации.
алкилароматических углеводородов необходимо вводить поправки, природа которых в основном имеет стерическое происхождение. Величина этих поправок никоим образом не является постоянной, как это принято, например, в методе Бенсона, а зависит от эффективных размеров взаимодействующих групп, от количества рядом расположенных заместителей и от их взаимной ориентации.
Последовательное применение метода Татевского по связям к накопленным к настоящему времени экспериментальным данным позволило определить значения парциальных вкладов, которые приведены в табл. 1.11. Все парциальные вклады получены на весьма представительных выборках и могут считаться достаточно надежными, чтобы быть рекомендованными к применению.
В отношении поправок (табл. 1.11) необходимо отметить, что большинство из них определено по одному-двум источникам экспериментальной информации. Однако все приведенные значения прошли дополнительное тестирование неэмпирическими методами расчета. Выполненный нами анализ полученных при этом результатов показал, что использование аддитивных подходов на этапе введения поправок для ![]() алкилароматических соединений, имеющих три и большее количество алкильных заместителей в молекуле, может рассматриваться лишь в качестве первого приближения. Недостаточно конструктивен, на наш взгляд, также подход, предложенный в свое время Коксом и Пилчером [2] для полизамещенных бензолов и состоящий в дополнении орто-эффектов поправками, учитывающими тройное взаимодействие заместителей в молекуле.
алкилароматических соединений, имеющих три и большее количество алкильных заместителей в молекуле, может рассматриваться лишь в качестве первого приближения. Недостаточно конструктивен, на наш взгляд, также подход, предложенный в свое время Коксом и Пилчером [2] для полизамещенных бензолов и состоящий в дополнении орто-эффектов поправками, учитывающими тройное взаимодействие заместителей в молекуле.
Таблица 1.11 Значения парциальных вкладов для прогнозирования ![]() *
*
Условное обозначение |
ΔfH0g, 298, кДж/моль | Условное обозначение | ΔfH0g, 298, кДж/моль | ||||||
| 1 уровень | 2 уровень | 3 уровень | N | 1 уро-вень | 2 уровень | 3 уровень | N | ||
| Парциальные вклады для ароматических соединений | |||||||||
| (Cb-H)1 | 13,877 | 65 | Cb-OH | -164,390 | 62 | ||||
| (Cb-C1)1 | -19,121 | 18 | Cb-NH2 | 18,42 | 16 | ||||
| (Cb-C2)1 | 13,976 | 10 | Cb-F | -181,85 | 34 | ||||
| (Cb-C3)1 | 24,824 | 11 | Cb-I | 93,424 | 6 | ||||
| (Cb-C4)1 | 34,603 | 13 | Nb | 70,45 | 12 | ||||
| (Cb-Cb)1 | 41,069 | 13 | |||||||
| Поправки на орто-взаимодействие заместителей | |||||||||
| C1-C1 (транс-“Н-Н”) | 1,461 | 15 | OH(транс-)-C2 | 2,4 | 1 | ||||
| C1-C1 (шахм.-“Н-Н”) | 5,120 | 6 | OH(транс-)-C3 | 2,029 | 6 | ||||
| C1-C2 | 3,600 | 1 | OH(цис-)-C3 | 8,661 | 1 | ||||
| C1-C3 | 5,900 | 1 | OH(транс-)-C4 | 11,155 | 15 | ||||
| C1-C4 | 23,600 | 1 | OH(цис-)-C4 | 18,007 | 3 | ||||
| C2-C2 | 4,222 | 1 | NH2-C4 | 19,05 | 2 | ||||
| C2-C4 | 35 | F-F | 21,956 | 19 | |||||
| C3-C3 | 10,204 | 1 | Cl-F | 13,279 | 4 |
| C4-C4 | 93 | I-I | 9,521 | 1 | |
| C1-Ph | 5,962 | 1 | (Nb-C1)орто | -7,63 | 7 |
| (Nb-C1)пара | -3,71 | 4 |
* - значения ряда парциальных вкладов и поправок для расчета Df H0 g , 298 скорректированы по сравнению с первой редакцией пособия в связи с увеличением представительности выборки веществ, участвующих в определении параметра.
Дело в том, что различие в энергиях конформеров, обусловленных взаимной ориентацией атомов и групп соседних заместителей в ароматическом ядре, оказывается достаточным, чтобы учесть этот фактор при формировании аддитивной схемы. Суть вопроса становится понятной при конформационном анализе соединений с относительно несложным строением молекул, например метилбензолов (МБ), проведенного с помощью программы Gaussian 03W методом B3LYP/6-311G++2d,2p. Указанный метод дает для о-ксилола два сосуществующих конформера (“А” и “В”, рис. 1.2)
Один из них (“А”) характеризуется взаимным транс-расположением атомов водорода метильных групп, лежащих в плоскости ароматического ядра, в другом (“В”) атомы водорода метильных групп имеют шахматную ориентацию по отношению друг к другу. Различие в энергиях этих конформеров по результатам расчета составляет для ![]() 5,2 кДж/моль. Очевидно, что при таком соотношении в энергиях о-ксилол представлен при 298 К преимущественно конформером “А”, и эффект взаимодействия заместителей (орто-эффект типа “метил-метил” или C1-C1(транс-“Н-Н”)) для
5,2 кДж/моль. Очевидно, что при таком соотношении в энергиях о-ксилол представлен при 298 К преимущественно конформером “А”, и эффект взаимодействия заместителей (орто-эффект типа “метил-метил” или C1-C1(транс-“Н-Н”)) для ![]() составляет 2,76 кДж/моль. Но очевидно также и то, что указанное расположение метильных групп возможно только для двух соседних заместителей, третий и последующие заместители уже не могут иметь транс-ориентации атомов водорода. Выполненный нами конформационный анализ показал, что для 1,2,3-триМБ энергетически наиболее выгодным конформером является “С” (рис. 1.3), который для двух соседних групп имеет транс-ориентацию атомов водорода, лежащих в плоскости ароматического ядра, а третья метильная группа подстроена к соседней “шахматно”. Таким образом, суммарный эффект взаимодействия заместителей, дестабилизирующий молекулу 1,2,3-триМБ, составляет для
составляет 2,76 кДж/моль. Но очевидно также и то, что указанное расположение метильных групп возможно только для двух соседних заместителей, третий и последующие заместители уже не могут иметь транс-ориентации атомов водорода. Выполненный нами конформационный анализ показал, что для 1,2,3-триМБ энергетически наиболее выгодным конформером является “С” (рис. 1.3), который для двух соседних групп имеет транс-ориентацию атомов водорода, лежащих в плоскости ароматического ядра, а третья метильная группа подстроена к соседней “шахматно”. Таким образом, суммарный эффект взаимодействия заместителей, дестабилизирующий молекулу 1,2,3-триМБ, составляет для ![]() 7,63 кДж/моль (2,76+4,87), а величину 2,11 кДж/моль, равную 7,63-2·2,76 кДж/моль, можно было бы воспринять в рамках схемы Кокса-Пилчера [2] как дополнительный “тройной” эффект (сверх удвоенного орто-эффекта). Однако указанная ситуация в группе метилбензолов реализуется только в случае 1,2,3,5-тетраМБ, т.е. носит довольно частный характер. Расчет показывает, что для 1,2,3,4-тетраМБ и пента-МБ две группы транс-ориентированы, а остальные шахматно подстроены. В случае гекса-МБ все заместители имеют шахматную ориентацию.
7,63 кДж/моль (2,76+4,87), а величину 2,11 кДж/моль, равную 7,63-2·2,76 кДж/моль, можно было бы воспринять в рамках схемы Кокса-Пилчера [2] как дополнительный “тройной” эффект (сверх удвоенного орто-эффекта). Однако указанная ситуация в группе метилбензолов реализуется только в случае 1,2,3,5-тетраМБ, т.е. носит довольно частный характер. Расчет показывает, что для 1,2,3,4-тетраМБ и пента-МБ две группы транс-ориентированы, а остальные шахматно подстроены. В случае гекса-МБ все заместители имеют шахматную ориентацию.
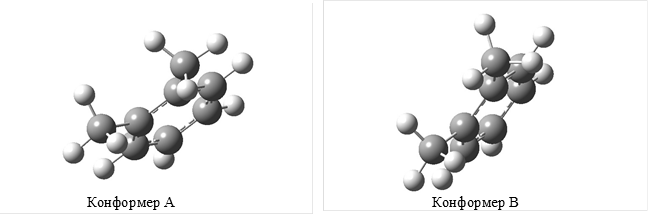
Рис. 1.2. Конформеры о-ксилола
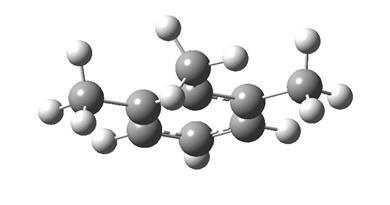
Рис. 1.3. Конформер C о-ксилола
На основании изложенного весь калориметрический материал для метилбензолов мог быть использован для определения всего двух поправок на орто-взаимодействие заместителей типа C1
-C1
(транс-“Н-Н”) и C1
-C1
(шахм.-“Н-Н”), значения которых приведены в табл. 1.11 и которые следует применять при прогнозировании ![]() целенаправленно в соответствии с взаимной ориентацией соседних групп.
целенаправленно в соответствии с взаимной ориентацией соседних групп.
Полагаем, что приведенный анализ и уровень значений орто-эффектов (табл. 1.11) предостережет от возможных ошибок при прогнозировании энтальпий образования веществ с более сложным строением молекул. Информация, представленная в табл. 1.11, свидетельствует о том, что спектр определенных орто-эффектов не охватывает всех возможных сочетаний взаимодействия алкильных заместителей и требуется пополнение экспериментальной базы. Для веществ со значительной степенью насыщения ароматического ядра алкильными заместителями различного строения приведенный набор параметров явно недостаточен. Однако детальный конформационный анализ интересующих структур с использованием возможностей методов молекулярной механики в сочетании со сведениями табл. 1.11 дает в большинстве случаев вполне удовлетворительные результаты при их оценке.
В отношении класса алкилфенолов в настоящее время можно сказать следующее. Введение всего одного парциального вклада типа “Cb-OH” (для учета взаимодействия ОН-группы с ароматическим ядром) в набор параметров, вычисленных на основе сведений по алкилбензолам и алканам, позволяет вполне корректно прогнозировать ![]() различных моно-, ди- и триметилфенолов, а также неэкранированных фенолов с алкильными заместителями иного строения. Частично экранированные и пространственно-затрудненные фенолы требуют учета эффектов взаимодействия ОН-группы с соседними алкильными заместителями. В методе Бенсона для этой цели рекомендована одна поправка, равная 2,38 и 1,42кДж/моль, в редакции [5] и [6] соответственно.
различных моно-, ди- и триметилфенолов, а также неэкранированных фенолов с алкильными заместителями иного строения. Частично экранированные и пространственно-затрудненные фенолы требуют учета эффектов взаимодействия ОН-группы с соседними алкильными заместителями. В методе Бенсона для этой цели рекомендована одна поправка, равная 2,38 и 1,42кДж/моль, в редакции [5] и [6] соответственно.
Значения орто-эффектов (табл. 1.11), определенные нами на основе экспериментальных данных, существенно зависят от эффективных размеров заместителей и их взаимной ориентации. Причем в случае фенолов последнее обстоятельство еще более четко выражено, чем для алкилбензолов. На основании многочисленных спектроскопических исследований и результатов выполненного нами конформационного анализа алкилфенолов (АФ) с различным строением молекул была установлена достаточно строгая ориентация гидрокси-групп по отношению как к плоскости ароматического ядра (практически всегда находится в плоскости), так и к соседнему алкильному заместителю. В соответствии с этим рекомендованы значения различных орто-эффектов, которые приведены в табл. 1.11 и предназначены для расчета ![]() с учетом следующего.
с учетом следующего.
При прогнозировании ![]() вторичных и третичных орто-алкилфенолов можно исходить из того, что АФ представлены одним конформером с транс-ориентацией атома водорода гидрокси-группы по отношению к соседнему алкильному заместителю. В молекулах 2,6-диалкилзамещенных фенолов реализуется и транс-, и цис-ориентация заместителей. В указанном приближении нами определены поправки для алкилфенолов, приведенные в табл. 1.11. В перечне орто-эффектов типа “ОН-Сi
” отсутствуют поправки типа “ОН-С1
” для орто- и 2,6-метилзамещенных фенолов, поскольку их значения оказались менее 1 кДж/моль.
вторичных и третичных орто-алкилфенолов можно исходить из того, что АФ представлены одним конформером с транс-ориентацией атома водорода гидрокси-группы по отношению к соседнему алкильному заместителю. В молекулах 2,6-диалкилзамещенных фенолов реализуется и транс-, и цис-ориентация заместителей. В указанном приближении нами определены поправки для алкилфенолов, приведенные в табл. 1.11. В перечне орто-эффектов типа “ОН-Сi
” отсутствуют поправки типа “ОН-С1
” для орто- и 2,6-метилзамещенных фенолов, поскольку их значения оказались менее 1 кДж/моль.
Среднее абсолютное отклонение расчетных значений ![]() от экспериментальных величин для всех рассмотренных фенолов составило 1,9 кДж/моль для метода Татевского и 6,9 кДж/моль для метода Бенсона при средней экспериментальной погрешности 1,8 кДж/моль. Таким образом, метод Татевского по связям показал достаточно высокую работоспособность при относительно небольшом наборе параметров.
от экспериментальных величин для всех рассмотренных фенолов составило 1,9 кДж/моль для метода Татевского и 6,9 кДж/моль для метода Бенсона при средней экспериментальной погрешности 1,8 кДж/моль. Таким образом, метод Татевского по связям показал достаточно высокую работоспособность при относительно небольшом наборе параметров.
В отличие от фенолов прогнозирование ![]() алкиланилинов не потребовало аналогичной детализации поправок на орто-взаимодействие заместителей, что объясняется различием в строении функциональных групп и их ориентации в молекуле. Для алкиланилинов оказывается достаточным дополнить рассматриваемую аддитивную схему одним парциальным вкладом типа ”Cb
-NH2
” и значением одного орто-эффекта типа “NH2
-C4
” для соседних амино-группы и алкильного заместителя с a-четвертичным атомом углерода. При этом важно отметить, что полученное значение указанного орто-эффекта практически совпадает с величиной эффекта типа “OH(цис-)
-C4
” и сохраняет свою величину при прогнозировании
алкиланилинов не потребовало аналогичной детализации поправок на орто-взаимодействие заместителей, что объясняется различием в строении функциональных групп и их ориентации в молекуле. Для алкиланилинов оказывается достаточным дополнить рассматриваемую аддитивную схему одним парциальным вкладом типа ”Cb
-NH2
” и значением одного орто-эффекта типа “NH2
-C4
” для соседних амино-группы и алкильного заместителя с a-четвертичным атомом углерода. При этом важно отметить, что полученное значение указанного орто-эффекта практически совпадает с величиной эффекта типа “OH(цис-)
-C4
” и сохраняет свою величину при прогнозировании ![]() 2,4,6-дитретбутиланилина. Для алкиланилинов с меньшими по сравнению с третбутильным эффективными размерами алкильного заместителя значения орто-эффектов типа “NH2
-Ci
” находятся в пределах погрешности эксперимента и потому не включены в набор параметров, приведенных в табл. 1.11. Среднее абсолютное отклонение расчетных значений
2,4,6-дитретбутиланилина. Для алкиланилинов с меньшими по сравнению с третбутильным эффективными размерами алкильного заместителя значения орто-эффектов типа “NH2
-Ci
” находятся в пределах погрешности эксперимента и потому не включены в набор параметров, приведенных в табл. 1.11. Среднее абсолютное отклонение расчетных значений ![]() алкиланилинов с использованием этих параметров составило 1,3 кДж/моль при средней погрешности эксперимента 1,7 кДж/моль. Метод Бенсона в редакции [5] дает среднее отклонение 3,8 кДж/моль.
алкиланилинов с использованием этих параметров составило 1,3 кДж/моль при средней погрешности эксперимента 1,7 кДж/моль. Метод Бенсона в редакции [5] дает среднее отклонение 3,8 кДж/моль.
Галогенбензолы
Энтальпии образования всех фторбензолов от моно- до гекса-замещенного требуют дополнения аддитивной схемы Татевского всего двумя параметрами: “Cb -F” и орто-эффектом типа “F-F”. При этом среднее абсолютное отклонение составляет 1,5 кДж/моль, метод Бенсона дает 13,6 кДж/моль.
Группа алкилфторбензолов, для которых известны ![]() , содержит всего три соединения, причем для 2-фтортолуола приведенные в [27] сведения представляются нам ошибочными. Таким образом, для выработки подходов к прогнозированию
, содержит всего три соединения, причем для 2-фтортолуола приведенные в [27] сведения представляются нам ошибочными. Таким образом, для выработки подходов к прогнозированию ![]() соединений этого класса нужна дополнительная информация.
соединений этого класса нужна дополнительная информация.
Для описания энтальпий образования от моно- до гексахлорбензола потребовалось введение в аддитивную схему, аналогично фторбензолам, всего двух параметров (табл. 1.11). При этом среднее абсолютное отклонение составляет 2,0 кДж/моль при средней погрешности эксперимента 2,7 кДж/моль, метод Бенсона дает 2,9 кДж/моль.
Экспериментальные данные по алкилхлорбензолам требуют существенного дополнения, для того чтобы можно было рекомендовать подход к прогнозированию их ![]() , особенно для орто-замещенных структур.
, особенно для орто-замещенных структур.
Группа фтор-хлор-бензолов с различным сочетанием заместителей, вплоть до полного насыщения ароматического ядра атомами галогена, потребовала дополнения набора параметров аддитивной схемы одним значением орто-эффекта типа “Cl-F”. Результаты прогнозирования ![]() всех веществ вполне удовлетворительны.
всех веществ вполне удовлетворительны.
Экспериментальные данные для иодбензолов имеют значительную погрешность и ограничены шестью структурами с несложным строением молекул. На основе этих данных определен парциальный вклад типа “Cb
-I” и выполнена оценка орто-эффекта типа “I-I”. Значения полученных величин приведены в табл. 1.11. Очевидно, что для выработки более аргументированного подхода к прогнозированию ![]() соединений этого класса требуется пополнение базы экспериментальными данными.
соединений этого класса требуется пополнение базы экспериментальными данными.
Полифенилы
Объем калориметрических данных для группы полифенилов весьма ограничен. На их основе можно говорить только о выработке подходов к прогнозированию ![]() голоядерных углеводородов. Для их описания в дополнение к информации, рассмотренной выше, оказывается достаточным знание одного парциального вклада типа “Cb
-Cb
”, характеризующего связь между ароматическими ядрами (табл. 1.11). Эффект взаимодействия соседних фенильных заместителей в ароматическом ядре оказался меньше погрешности эксперимента для полифенилов и принят равным нулю. Среднее абсолютное отклонение составляет 2,4 и 3,2 кДж/моль в методах Татевского и Бенсона соответственно. Сведения по 2-метилбифенилу дают величину орто-эффекта типа “метил-фенил”, равную 4,7 кДж/моль. Это позволяет сделать вывод о том, что алкильные заместители с большим эффективным объемом, чем в случае метильной группы, вызовут существенную дестабилизацию молекулы при их нахождении в положениях 2, 2' и аналогичных им. Очевидно также, что для алкилзамещенных полифенилов, как и для алкилбензолов, не может использоваться единое значение орто-эффекта, каким бы оно ни было.
голоядерных углеводородов. Для их описания в дополнение к информации, рассмотренной выше, оказывается достаточным знание одного парциального вклада типа “Cb
-Cb
”, характеризующего связь между ароматическими ядрами (табл. 1.11). Эффект взаимодействия соседних фенильных заместителей в ароматическом ядре оказался меньше погрешности эксперимента для полифенилов и принят равным нулю. Среднее абсолютное отклонение составляет 2,4 и 3,2 кДж/моль в методах Татевского и Бенсона соответственно. Сведения по 2-метилбифенилу дают величину орто-эффекта типа “метил-фенил”, равную 4,7 кДж/моль. Это позволяет сделать вывод о том, что алкильные заместители с большим эффективным объемом, чем в случае метильной группы, вызовут существенную дестабилизацию молекулы при их нахождении в положениях 2, 2' и аналогичных им. Очевидно также, что для алкилзамещенных полифенилов, как и для алкилбензолов, не может использоваться единое значение орто-эффекта, каким бы оно ни было.
Для дифенил-, трифенил- и тетрафенилметанов методом Бенсона в редакции [5] предусмотрены три самостоятельных парциальных вклада для центральных атомов углерода. Метод Татевского позволяет достаточно корректно описывать ![]() указанных соединений при дополнении парциальных вкладов (табл. 1.11), полученных на основе алкилбензолов, всего одной поправкой типа “(Cb
-Cb
)2
”, учитывающей взаимное влияние двух ароматических ядер, разделенных двумя связями. Результаты прогнозирования показывают, что наличие заместителей в положениях “2” ароматических ядер может потребовать внесения в схему дополнительных корректив, особенно в тех случаях, когда заместители объемны.
указанных соединений при дополнении парциальных вкладов (табл. 1.11), полученных на основе алкилбензолов, всего одной поправкой типа “(Cb
-Cb
)2
”, учитывающей взаимное влияние двух ароматических ядер, разделенных двумя связями. Результаты прогнозирования показывают, что наличие заместителей в положениях “2” ароматических ядер может потребовать внесения в схему дополнительных корректив, особенно в тех случаях, когда заместители объемны.
В отношении фтор-, хлор- и бром-бифенилов можно с удовлетворением отметить достаточно высокое качество экспериментальных данных и наличие их объема, необходимого для ответа на принципиально важные вопросы прогнозирования ![]() . Для всех соединений, за исключением 2-, 2,2'- и 2,6-галогензамещенных бифенилов, энтальпии образования вполне удовлетворительно описываются набором параметров, приведенных в табл. 1.11, т.е. в молекулах отсутствуют значимые энтальпийные эффекты в общей цепи сопряжений с участием ароматических ядер. Интересен и тот факт, что изменение природы и размеров атомов галогена практически не изменило величины энтальпийной составляющей орто-эффекта для 2-, 2,2'- и 2,6-замещеных бифенилов. Это говорит о малой чувствительности рассматриваемого свойства к изменению размеров двугранного угла между ароматическими ядрами при переходе от фтор- к бром-производным бифенила.
. Для всех соединений, за исключением 2-, 2,2'- и 2,6-галогензамещенных бифенилов, энтальпии образования вполне удовлетворительно описываются набором параметров, приведенных в табл. 1.11, т.е. в молекулах отсутствуют значимые энтальпийные эффекты в общей цепи сопряжений с участием ароматических ядер. Интересен и тот факт, что изменение природы и размеров атомов галогена практически не изменило величины энтальпийной составляющей орто-эффекта для 2-, 2,2'- и 2,6-замещеных бифенилов. Это говорит о малой чувствительности рассматриваемого свойства к изменению размеров двугранного угла между ароматическими ядрами при переходе от фтор- к бром-производным бифенила.
Практически незначим и эффект взаимодействия заместителей в молекуле 2-фениланилина, о чем свидетельствуют результаты расчета его ![]() , полученные без учета орто-эффекта типа “NH2
-Ph”.
, полученные без учета орто-эффекта типа “NH2
-Ph”.
Пиридины
Сведения по алкилпиридинам ограничены метилпроизводными. На их основе можно ответить только на принципиальные вопросы прогнозирования ![]() . Результаты расчета показывают, что значимыми являются эффекты орто- и пара-взаимодействия заместителей в молекуле. Таким образом, для метилпиридинов набор парциальных вкладов (табл. 1.11) дополнен величиной парциального вклада для атома азота ароматического ядра (Nb
) и двумя значениями эффектов типа “(Nb
-C1
)орто” и “(Nb
-C1
)пара”. Среднее абсолютное отклонение расчетных величин с использованием этих параметров составило 1,1 кДж/моль для рассмотренной нами выборки веществ при средней погрешности эксперимента 0,6 кДж/моль. Метод Бенсона для той же выборки дает 2,2 кДж/моль. Из приведенной информации очевидно, что для алкилпиридинов с заместителями иного строения необходимо пополнение базы данных экспериментальными сведениями.
. Результаты расчета показывают, что значимыми являются эффекты орто- и пара-взаимодействия заместителей в молекуле. Таким образом, для метилпиридинов набор парциальных вкладов (табл. 1.11) дополнен величиной парциального вклада для атома азота ароматического ядра (Nb
) и двумя значениями эффектов типа “(Nb
-C1
)орто” и “(Nb
-C1
)пара”. Среднее абсолютное отклонение расчетных величин с использованием этих параметров составило 1,1 кДж/моль для рассмотренной нами выборки веществ при средней погрешности эксперимента 0,6 кДж/моль. Метод Бенсона для той же выборки дает 2,2 кДж/моль. Из приведенной информации очевидно, что для алкилпиридинов с заместителями иного строения необходимо пополнение базы данных экспериментальными сведениями.