| Скачать .docx |
Реферат: Диэлектрическая релаксация и подвижность мезогенных групп в холестеринсодержащих жидкокристаллических полимерах с развязками различной длины
ДИЭЛЕКТРИЧЕСКАЯ РЕЛАКСАЦИЯ И ПОДВИЖНОСТЬ МЕЗОГЕННЫХ ГРУПП В ХОЛЕСТЕРИНСОДЕРЖАЩИХ ЖИДКОКРИСТАЛЛИЧЕСКИХ ПОЛИМЕРАХ С РАЗВЯЗКАМИ РАЗЛИЧНОЙ ДЛИНЫ
По сравнению с низкомолекулярными аналогами мезогенные группы, присоединенные к полимерной цепи, в движении, необходимом для реализации ЖК-состояния, испытывают ограничения, связанные с наличием ковалентных связей с цепью. В результате оказываются измененными стерические условия, усиливается затрудненность внутримолекулярного вращения, причем очевидно, что молекулярная подвижность мезогенных групп будет зависеть еще и от релаксационного состояния, в котором при данной температуре находится полимер — стеклообразного, высокоэластического и т. п. В связи с этим при рассмотрении релаксационного поведения ЖК-полимеров прямой перенос представлений о вращении мезогенного фрагмента как целого по аналогии с низкомолекулярными жидкими кристаллами, по-видимому, недопустим. Возникает вопрос о механизмах образования ЖК-порядка в полимерах и о сходстве или различиях способов его реализации в низкомолекулярных соединениях и полимерах.
Мезофазные переходы (образование, изменение и плавление мезоморфных областей) могут осуществляться только вследствие подвижности мезогенных групп, достаточной для прохождения требуемой перестройки в их пространственном расположении. В отличие от низкомолекулярных веществ, где такое пространственное подстраивание реализуется при движении молекул как целого, в полимерах источником необходимых пространственных смещений является главным образом поворотная изомерия, т. е. внутреннее вращение локальных группировок относительно простых ковалентных связей.
Интерпретация релаксационных процессов требует учета специфики молекулярного движения в полимерах, которая состоит в множественности внутрицепных форм подвижности. Можно ожидать, что для полимеров с мезогенными группировками в боковых цепях движение сегментов основного хребта макромолекулы, которое отличается высокой неоперативностью и наличием внутри- и межцепных корреляций, будет оказывать влияние на подвижность мезогенных групп, ослабевающее при введении в боковую цепь кинетических развязок.
Кинетической автономии мезогенные группы могут достичь при отделении их от главной цепи линейной цепочкой, например полиметиленовой последовательностью. Можно предположить, что оптимальная длина развязки будет определяться строением как главной цепи, так и самих мезо-генных групп.
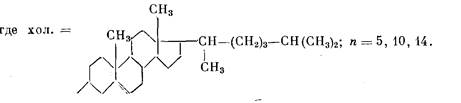
Цель настоящей работы — анализ молекулярной подвижности мезотенных групп, обеспечивающей мезофазные переходы, и изучение влияния длины полиметиленовой цепочки в боковой цепи, отделяющей мезогенный фрагмент от основной цепи, на локальную подвижность мезогенных групп. Исследования проводили на образцах холестеринсодержащих полимеров (ПХМ-и) общей формулы [1]
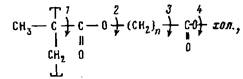
ПХМ-14 частично исследовали ранее [2]. Здесь приведены дополнительные сведения, касающиеся особенностей молекулярного движения при низких температурах, а также релаксационного поведения, связанного с ЖК-состоянием полимера. Суждения о молекулярном движении вынесены на основе изучения релаксации дипольной поляризации рассматриваемых систем. Основную часть измерений температурно-частотных зависимостей tg б (б — угол диэлектрических потерь) и электрической емкости образцов выполняли в диапазоне частот от 0.06 до 300 кГц с помощью прибора TR-9701 при температурах от — 170 до 230°. Образцы готовили прессованием выше температуры стеклования либо отливом пленок из раствора в толуоле. Толщина образцов 20—50 мкм.
В работе [1] показано, что все исследованные здесь полимеры образуют ЖК-структуру, что обусловлено наличием мезогенных холестериновых групп. При изучении этих полимеров диэлектрическим методом существенно, что каждое монозвено содержит две сложноэфирные группы, разделенные гибкой развязкой из п метиленовых групп. Поэтому можно ожидать, что релаксация их диполыюй поляризации будет проявляться независимым образом и с разными временами, так как ближайшее их окружение и взаимодействия неравноценны. Как было показано в работе [2], вторая сложноэфирная группа, примыкающая к холестериновому радикалу, кор-релирована в своем движении с холестериновой группой.
Наличие двух полярных групп приводит к тому, что при низких температурах для всех изученных полимеров наблюдали три области прохождения tg б через максимум (рис. 1). Координаты самой низкотемпературной области релаксации (—120°) близки к таковым для акрилатного и метакрилатного рядов полимеров гребнеобразного строения [3]. Эту область диэлектрических потерь можно связать с подвижностью сложно-эфирных групп, смежных с основной цепью, и примыкающих к ним нескольких групп СН2 боковой полиметиленовой цепочки.
Вторая релаксационная область диэлектрических потерь вблизи — 50° связана с локальной подвижностью второй сложноэфирной группы, расположенной при холестериновом радикале. На это указывают данные по диэлектрической релаксации в гребнеобразных полимерах с кислотными фрагментами, в которых полярная группа находится в тех же условиях, что и в ПХМ-n, и где эта область при —50° по величине tg бмакс в сильной степени зависит от присутствия в образце влаги [4].
Такое соотнесение низкотемпературных процессов релаксации дипольной поляризации с локальными формами движения обеих сложноэфирных групп согласуется также с результатами изучения дипольных явлений в полимерах аналогичного строения в растворе. При введении в боковые цепи гребнеобразного полимера второй полярной группы, отделенной от
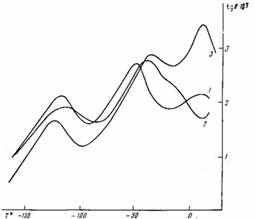
Рис. 1. Температурные зависимости tgб для ПХМ-5, ПХМ-10 (2) и ПХМ-14 (3) при 0,1 кГц (области низкотемпературных процессов) первой достаточно гибкой развязкой, появляется второй процесс релаксации, свидетельствующий о локальном характере и автономии движения обеих сложноэфирных групп [5].
Третья область диэлектрических потерь (вблизи —15° для ПХМ-5 и 10—15° для ПХМ-5 и ПХМ-14) в рассматриваемых полимерах также должна быть отнесена к локальному движению в пределах боковой цепи, включающему вторую группу СОО при холестериновом радикале. Это следует из того, что в гребнеобразных полимерах акрилатных и метакрилатных рядов такого процесса нет [3], хотя пока однозначная интерпретация механизмов данного процесса преждевременна.
Все три вида поляризации имеют невысокую энергию активации (33, 41—50 и 60 кДж/моль), что также указывает на значительную кинетическую гибкость боковых цепейПХМ-5, ПХМ-10и ПХМ-14.
По сравнению с гребнеобразными полимерами акрилатного и метакрилагного рядов, не имеющими мезогенпых групп в боковых цепях, данныесистемы отличает существование специфических областей диэлектрических потерь в области более высоких температур. Как следует из рис. 2, для всех трех полимеров наблюдается переход в изотропное состояние: при 210, 153 и 150° для ПХМ-5, ПХМ-10 и ПХМ-14 соответственно tg б проходит через не зависящий от частоты максимум, имеющий асимметричную форму, а в случае ПХМ-5 для зависимости tg б от температуры появляется характерное плечо. Температура этого перехода совпадает с данными, полученными с помощью поляризационного микроскопа [1].
Кроме перехода, обусловленного плавлением ЖК-фазы, в работе [1] для рассматриваемых здесь систем методом ДСК обнаружены мезоморфные переходы, связанные с изменением смектического порядка. Они наблюдались при 190, 124 и 54° для ПХМ-5, ПХМ-10 и ПХМ-14 соответственно. Указанные переходы проявляются и в температурных зависимостях диэлектрических потерь, хотя и с разной степенью отчетливости. Наиболее очевиден и воспроизводится при всех повторных измерениях и на всех образцах не зависящий от частоты пик tg б при 190° в ПХМ-5 (рис. 2). Менее отчетливо, в виде некоторого плеча, обозначен такого рода переход вблизи 55° в ПХМ-14 (рис. 2, в).
В ПХМ-10 температурное положение промежуточного мезофазного перехода (124°) совпадает с пиком диэлектрических релаксационных потерь (ниже он будет обозначен цифрой 1) при высоких частотах (рис. 2, б). Наблюдаемую при этих температурах область tg 6MaK c отличает значительная размазанность по температуре, что можно рассматривать как следствие наложения релаксационного процесса и мезоморфного структурного перехода. Естественно, что такое заключение может быть сделано только при наличии прямых данных ДСК [1].
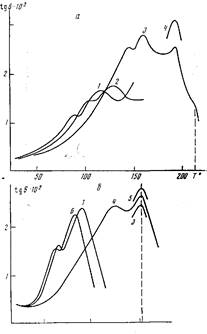
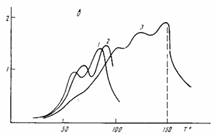
Рис. 2. Температурные зависимости tg б для ПХМ-5 (а), ПХМ-10 (б) и ПХМ-14 (в) при частотах 0,1 {1), 1 (2), 100 (5), 300 (4), 500 (5) и 0,03 Гц {6)
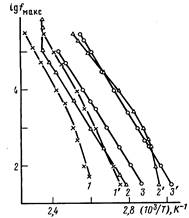
Рис. 3. Зависимость lg/максот обратной температуры для релаксационных процессов I (1-3) и II Ц'-З') в ПХМ-5 (1, Г), ПХМ-10 {2, 2') и в ПХМ-14 (3, 3')
Для каждого из исследованных полимеров обнаружены еще две области релаксационных потерь (I и II). Соответствующие зависимости частоты максимума tgб от обратной температуры (1gfмакс =φ(1/Г)),приведены на рис. 3. Значения энергии активации и температуры процесса, определенной путем экстраполяции температурно-частотных координат к 1 Гц, приведены в таблице. Здесь же даны температуры стеклования Тс , заимствованные из работы [1].
Оба релаксационных процесса близки по температуре, являются кооперативными, так как их энергия активации велика (для локальных она не превышает 33—62 кДж/моль).
Для ПХМ-5 оба процесса (I и II), а для ПХМ-10 — только низкотемпературный (I) обнаруживают искривление зависимости 1gfмакс =φ(1/Г), что раньше отмечали для дипольно-сегментальных переходов при температурах на 50—100° выше Тс . Если для ПХМ-5 процесс II более заторможен (энергия активации и температура выше), чем I, то для ПХМ-10 и ПХМ-14 скорее наоборот, энергия активации оказалась ниже для процесса II, хотя соответствующие ему температуры по-прежнему более высоки.
Для ПХМ-10 установлено, что процесс II имеет место вплоть до температуры просветления и практически переходит в структурный, сопровождающий плавление ЖК-фазы.
Можно предположить, что такие закономерности, как кривизна зависимости процессов I и II по температурно-частотным координатам (показано для ПХМ-5 на рис. 3) и затем диэлектрическое проявление перехода из ЖК-состояния в изотропное, являются характерными для полимеров с мезогенными группами в боковых цепях. Подобного рода наблюдения были сделаны для полимеров с силоксановым хребтом макроцепи [6—8], для полиакрилатов и метакрилатов с другими типами мезогенных групп-[9,10].
В работах [6—8] происхождение процессов с признаками, аналогичными процессам I и II данной работы, связывали с ориентационными поворотами анизодиаметрической мезогенной группы, в результате которых происходила ориентация параллельной μ и перпендикулярной μх составляющих дипольного момента мезогенной группы. Предположение основано на перенесении механизмов ориентационных процессов в низкомолекулярных жидких кристаллах на мезогенные фрагменты, введенные в полимерные цепи. Эта трактовка может быть использована и для интерпретации сходных переходов в полимерах с холестериновыми мезогенными группами в боковых цепях.
Полярной группой, определяющей и μ и μ± , является группа СОО, примыкающая к холестериновому радикалу. Выше было показано, что она является источником происхождения процесса с очень малыми временами (при низких температурах). Объемный холестериновый радикал в части, примыкающей к СОО, практически лишен внутренних движений и потому, учитывая малые времена, в низкотемпературном локальном движении не участвует. Можно считать, что кинетическая единица низкотемпературного процесса ограничена, с одной стороны, вращением СОО относительно связи 4, а с другой — примыкающими метиленовыми группами из числа (СН2 ).
Участие холестеринового радикала в процессе I доказано путем сопоставления диэлектрических и ЯМР данных для холестеринового эфира поли-метакрилоил-со-аминолауриновой кислоты, а по аналогии и для ПХМ-n. Можно полагать, что процесс I, располагающийся при более низких температурах, следует отождествить с ориентацией μ± второй группы СОО в совокупности с холестериновым радикалом, простейший путь которой осуществляется при вращении относительно связи 3.
Несколько труднее интерпретировать молекулярную подвижность, обусловливающую появление процесса II, лежащего при более высоких температурах. Его характеристики подходят под определение ориентационного движения μ IIмезогенной группы. Кроме того, совпадение его температур с температурой стеклования (таблица), сдвиг области диэлектрических потерь к низким температурам при введении пластификатора [2], высокие значения энергии активации придают сходство с тривиальными дипольно-сегментальными процессами (α-переход), характерными для аморфных полимеров в области стеклообразования.
При рассмотрении представленной альтернативы, по-видимому, полезно учесть дополнительное обстоятельство, отмеченное в работе [11]. Исследуя диэлектрическую релаксацию в сополимерах 1-метакрнлоилокси-бензоил-феннлен-4-анисоата со стиролом, было установлено, что α-переход, связанный со стеклованием, наблюдали только в системах, где количественный состав соответствовал получению аморфных изотропных образцов. Этот процесс исчезал, как только концентрация мезогенного компонента становилась достаточной для реализации ЖК-состояния. α -Переход диэлектрическим методом отчетливо не наблюдался и в ряде других гребнеобразных полимеров, например в поли-1М-октадецилакрил- и метакриламн-дах [3]. В связи с этим можно предположить, что процесс II, лежащий на 20—25 выше, чем процесс I, с близкими для обоих процессов энергиями активации, является следствием ориентациоиного движения μ//. Для осуществления этого движения достаточно предположить наличие внутрицеп-ной корреляции в пределах холестериновый радикал — группа СОО и по крайней мере одна прилежащая группа СН2 . Вращение такой последовательности относительно связей, входящих в полиметиленовую цепочку, обеспечивает изменение ориентации μ//и необходимую пространственную подстройку в реализации ЖК-состояния.
Можно считать, что внутреннее вращение в кинетически гибких цепях, несущих мезогенные фрагменты, способно обеспечить ориентационное движение, характерное для низкомолекулярных жидких кристаллов с анизодиаметричными мезогенными группами. По-видимому, близость координаттакого рода движения к температуре стеклования не случайна, так как движение при этом совершают протяженные участки с корреляцией внутреннего вращения как вдоль боковой цени, так и межцепной (между боковыми привесками), что и объясняет высокие значения активационных величин.
Представления об отнесении процессов I и II в полимерах с мезогенными группами в боковых цепях нуждаются в доказательстве общности этого явления, так как два процесса с признаками, подобными описанным, наблюдались в полимерах с азометиновыми группировками, где перпендикулярная составляющая дипольного момента незначительна и вряд ли может обеспечить столь четко выраженную область диэлектрических потерь, как это наблюдалось в эксперименте [12].
В заключение рассмотрим влияние длины кинетической развязки на проявление описанных закономерностей, связанных с процессами I и II. Как видно из рис. 3, для полимера с развязкой из пяти групп СН2 характерна не только самая высокая температура просветления, но и более высокие температуры и активационные величины процессов I и II. Заторможенность движения соответствующих кинетических единиц эффективно снижается прп переходе к n=10. Дальнейшее удлинение развязки (n=14) существенных изменений в параметрах релаксации не вызывает, можно лишь отметить некоторое сближение процессов.
Таким образом, можно сделать вывод о том, что реализация ЖК-состояния в полимерах, где боковой привесок несет в себе мезогенный фрагмент, определяется условиями внутреннего вращения в боковых цепях, которое способно обеспечить повороты мезогенной группы относительно как продольных, так и поперечных осей.
СПИСОК ЛИТЕРАТУРЫ
1. Freidzvn Ya. S., Kharitonov Л. V., Shibaev V. P., Plate N. A. // Advances in liquidcrystal research and applications/Ed. by Bata L., Budapest. 1980. V. 2. P. 899.
2. Борисова Т. И., Бурштейн Л. Л., Никонорова И. А., Фрейдзоп Я. С, Шибаев В. П., Плат.) Н. А. II Высокомолек. соед. А. 1984. Т. 26. № 7. С. 153.
3. Борисова Т. П., Бурштейн Л. Л., Никонорова И. А., Шибаев В. П. // Высокомолек. соед. А. 1982. Т. 24. № 8. С. 1669.
4. Тальрсзе Р. В., Нараханова Ф. И., Борисова Т. И., Бурштейн Л. Л., Никонорова И. А., Шибаев В. П., Платэ Н. А. // Высокомолек. соед. А. 1978. Т. 20. № 8. С. 1835.
5. Борисова Т. П.. Бурштейн Л. Л., Степанова Т. П., Харитонов А. В., Фрейдзон Я. С, Шибаев В. U. II Высокомолек. соед. А. 1982. Т. 24. № 7. С. 1463.
6.Atlard f. S., Williams G.. Gray G. W., Lacey D., Gemmel P. A. // Polymer. 1986. V. 27. Д2. P. 185.
7. Atlard G. S., Williams G. II Polymer Commims. 1986. V. 27. № 1. P. 2.
8. Atlard C. S. Williams G. II Polymer Commims. 1986. V. 27. № 2. P. 66.
9. Zentel R.. Strobl G.. Rindsdorj H. // Macromolecules. 1985. V. 18. № 5. P. 900.
10. Ringsdorf H., Zentel R. // Makromolok. Chcm. 1982. B. 183. S. 1245.
11. Никонорова H. А., Малиновская В. П., Порисова Т. И., Бурштейн Л. Л., Коршун А. М., Скороходов С. С. II Высокомолек. соед. А. 1987. Т. 29. № 3. С. 549.
12. Борисова Т. И., Бурштейн Л. Л., Никонорова Н. А., Тальрозе Р. В., Шибаев В. П. //Высокомолек. соед. А. 1986. Т. 28. № 11. Р. 2335.