| Скачать .docx |
Курсовая работа: Содержание тяжелых металлов в пробах снега в зоне влияния Кирово-Чепецкого химического комбината
МИНОБРНАУКИ РОССИИ
ВЯТСКИЙ ГОСУДАРСТВЕННЫЙ ГУМАНИТАРНЫЙ УНИВЕРСИТЕТ
Химический факультет
Кафедра химии
Курсовая работа на тему:
«Содержание тяжелых металлов в пробах снега в зоне влияния Кирово-Чепецкого химического комбината»
Выполнил:
студент 4 курса
Кулябин Александр Николаевич
Научный руководитель:
доцент кафедры химии, к.б.н.
Скугорева Светлана Геннадиевна
Киров, 2011
Содержание
1. Введение
2. Задачи
3. Глава I
3.1. Метод атомно-абсорбционной спектрометрии
3.2. Метод вольтамперометрии
4. Глава II
4.1. Пробоотбор
4.2. Методика проведения химического анализа
4.2.1. Методика атомно-абсорбционной спектрометрии и обработка результатов
4.2.2. Методика вольтамперометрии и обработка результатов
5. Глава III
5.1. Результаты и их обсуждение
5.2. Заключение
6. Список литературы.
Введение:
1. Актуальность.
Присутствие кадмия в водной среде обусловлено его поступлением вследствие выщелачивания горных пород и почв, а также со сточными водами предприятий горнодобывающей, металлургической и химической промышленности. В незагрязненных речных и озерных водах кадмий присутствует в низких концентрациях - доли и единицы микрограммов в кубическом дециметре.
В поверхностных водах суши соединения кадмия находятся в растворенном и взвешенном состоянии. В состав взвеси входят, как правило, сорбированные формы. В растворенном состоянии кадмий существует в ионной форме, а также в виде неорганических и органических комплексов. В кислых водах преобладающей является наиболее токсичная ионная форма кадмия.
2. Действие на окружающую среду и человека:
Кадмий обнаруживается в организмах практически всех животных (у наземных около 0,5 мг на 1 кг массы, а у морских - от 0,15 до 3 мг/кг). Вместе с тем его относят к наиболее токсичным тяжелым металлам. Кадмий сосредотачивается в организме преимущественно в почках и печени, при этом содержание кадмия в организме к старости повышается. Он накапливается в виде комплексов с белками, которые участвуют в ферментативных процессах. Попадая в организм извне, кадмий оказывает ингибирующее действие на целый ряд ферментов, разрушая их. Его действие основано на связывании группы -SH цистеиновых остатков в белках и ингибировании SH-ферментов. Он может также ингибировать действие цинксодержащих ферментов, замещая цинк. Из-за близости ионных радиусов кальция и кадмия, он может замещать кальций в костной ткани.
Люди отравляются кадмием, употребляя воду, загрязненную кадмиесодержащими отходами, а также овощи и зерновые, растущие на землях, расположенных вблизи от нефтеперегонных заводов и металлургических предприятий. Особой способностью накапливать кадмий отличаются грибы. По некоторым сведениям, содержание кадмия в грибах может достигать единиц, десятков и даже 100 и более миллиграммов на кг собственной массы. Соединения кадмия есть среди вредных веществ, находящихся в табачном дыме (одна сигарета содержит 1-2 мкг кадмия).
Классическим примером хронического отравления кадмием является заболевание, впервые описанное в Японии в 1950-е и получившее название «итай-итай». Болезнь сопровождалась сильными болями в поясничной области, болью в мышцах. Появлялись и характерные признаки необратимого поражения почек. Были зафиксированы сотни смертельных исходов «итай-итай». Заболевание приняло массовый характер в силу высокой загрязненности окружающей среды в Японии в то время и специфики питания японцев - преимущественно рисом и морепродуктами (они способны накапливать кадмий в высоких концентрациях). Исследования показали, что заболевшие «итай-итай» потребляли до 600 мкг кадмия в сутки. В дальнейшем в результате мероприятий по охране окружающей среды, частота и острота синдромов, подобных «итай-итай» заметно снизилась.
В США была обнаружена зависимость между содержанием кадмия в атмосфере и частотой смертельных случаев от сердечно-сосудистых заболеваний.
Считают, что без вреда для здоровья в организм человека в сутки может поступать около 1 мкг кадмия на 1 кг собственного веса. В питьевой воде кадмия не должно содержаться более 0,01 мг/л. Противоядием при отравлении кадмием является селен, однако употребление продуктов, богатых этим элементом, приводит к понижению содержания серы в организме, и в этом случае кадмий снова становится опасным.
Основные источники кадмия - промежуточные продукты цинкового производства. Осадки металлов, полученные после очистки растворов сульфата цинка действием цинковой пыли, содержат 2-12% кадмия. Во фракциях, образующихся при дистилляционном получении цинка, содержится 0,7-1,1%о кадмия, а во фракциях, полученных при ректификационной очистке цинка - до 40%> кадмия. Кадмий извлекают и из пыли свинцовых и медеплавильных заводов (она может содержать до 5% и 0,5%о кадмия, соответственно). Пыль обычно обрабатывают концентрированной серной кислотой, а затем сульфат кадмия выщелачивают водой.
Задачи
· Проанализировать особенности методов определения тяжелых металлов: метода инверсионной вольтамперометрии и атомно-абсорбционной спектрометрии
· Провести пробоподготовку проб снега к анализу методом инверсионной-вольтамперомтерии
· Осуществить анализ проб на содержание тяжелых металлов методом инверсионной-вольтамперометрии
· Обработать и обсудить полученные данные
· Сравнить данные, полученные вольтамперомтерическим методом с результатами полученными на атомно-абсорбционном спектрометре
· Сформулировать выводы по проделанной работе
Глава I. Обзор методов
1. Метод вольтамперометрии
Вольтамперометрия- совокупность электрохимических методов исследования и анализа, основанных на изучении зависимости силы тока в электролитической ячейке от потенциала погруженного в анализируемый раствор индикаторного микроэлектрода, на котором реагирует исследуемое электрохимически активное (электроактивное) вещество. В ячейку помещают помимо индикаторного вспомогательный электрод со значительно большей поверхностью, чтобы при прохождении тока его потенциал практически не менялся (неполяризующийся электрод). Разность потенциалов индикаторного и вспомогательного электродов Е описывается уравнением Е = U— IR, где U- поляризующее напряжение, R - сопротивление раствора, I - ток в электохимической ячейке. В анализируемый раствор вводят в большой концентрации индифферентный фоновый электролит, чтобы, во-первых, уменьшить величину Rи, во-вторых, исключить миграционный ток, вызываемый действием электрического поля на электроактивные вещества. При низких концентрациях этих веществ изменение омического сопротивления электролита в ячейке и падения напряжения IRв растворе очень мало. Для полной компенсации изменения омического сопротивления и падения напряжения применяют потенциостатирование и трехэлектродные ячейки, содержащие дополнительно электрод сравнения.
В качестве индикаторных электродов используют стационарные и вращающиеся электроды - из металла (серебро, золото, платина), углеродных материалов (графит), а также капающие электроды (из ртути, амальгам, галлия). Последние представляют собой капилляры, из которых по каплям вытекает жидкий металл.
Вольтамперометрия с использованием капающих электродов, потенциал которых меняется медленно и линейно, называют полярографией. Электродами сравнения служат обычно электроды второго рода, например каломельный или хлоросеребряный.
Циклическая вольтамперометрия (вольтамперометрия с относительно быстрой треугольной разверткой потенциала) позволяет изучать кинетику и механизм электродных процессов путем наблюдения на экране компьютера или диаграмме потенциометра вольтамперограмм, отражающих концентрации анализируемых компонентов и электрохимические реакции продуктов электролиза.
Для всех вариантов вольтамперометрии используют способ снижения Сn , основанный на предварительном электрохимическом, адсорбционном или химическом накоплении определяемого компонента раствора на поверхности или в объеме стационарного микроэлектрода, с последующей регистрацией вольтамперограммы, отражающей электрохимическую реакцию продукта накопления. Эту разновидность вольтамперометрииназывают инверсионной. Винверсионной вольтамперометрии с предварительным накоплением, достигает 10-9 -10-11 . Минимальные значения Сn получают, используя углеродные и тонкопленочные ртутные индикаторные электроды, в т.ч. ртутно-графитовые, состоящие из мельчайших капелек ртути, электролитически выделенных на подложку из специально обработанного графита.
Вольтамперометриюприменяют:
• для количественного анализа неорганических и органических веществ в очень широком интервале содержаний - от 10-10 % до десятков %;
• для исследования кинетики и механизма электродных процессов, включая стадию переноса электрона, предшествующие и последующие химической реакции, адсорбцию исходных продуктов и продуктов электрохимических реакций и т. п.;
• для изучения строения двойного электрического слоя, равновесия комплексообразования в растворе, образования и диссоциации интерметаллических соединений в ртути и на поверхности твердых электродов; для выбора условий амперометрического титрования и др. .
•
2. Атомно-абсорбционная спектрометрия
Атомно-абсорбционные спектрометры выпускаются с атомизаторами различного типа:пламенными и электротермическими.
Схема спектрометра, использующего пламенный атомизатор, представлена на рис. 1. Такой спектрометр состоит из источника излучения (1), атомизатора - пламени (2), монохроматора (3) и детектора - приемника света (4), также имеется двухлинзовая оптическая система (5).
|

Рис.1. Схема атомно-абсорбционного спектрометра с пламенной атомизацией пробы. |
При анализе методом атомной абсорбции в спектрометрах с разными видами атомизаторов в качестве источника первичного излучения линейчатого спектра часто используют лампы с полым катодом (ЛПК), содержащим определяемый элемент. Лампа с полым катодом представляет собой цилиндрический стеклянный баллон с кварцевым или стеклянным окошком, заполненный аргоном или неоном (давление ~ 102 Па), в котором происходит испарение вещества и возбуждение атомов элемента при электрическом заряде в атмосфере инертного газа. Лампа с полым катодом испускает интенсивные узкие линии элемента, входящего в состав катода.
Анод такой лампы - металлическая вольфрамовая проволока, находящаяся рядом с катодом. Катод представляет собой полый цилиндр, изготовленный из определяемого элемента или его сплава. Катод и анод размещены в стеклянном цилиндре. Когда на электроды лампы подается напряжение от высокоточного выпрямителя около 600 В, газ ионизируется. Катионы газа выбивают из катода атомы определяемого элемента и возбуждают их термически. При обратном переходе возбужденных атомов в основное состояние излучается свет определенных длин волн. В спектре свечения при температуре 800 К в полом катоде наблюдаются резонансные частоты элемента.
Металл, используемый для изготовления ламп с полым катодом, должен быть высокой чистоты и не содержать адсорбированный водород. Работу лампы ухудшается из-за снижения давления газа вследствие частичной его сорбции на катоде.
Для атомизации в атомно-абсорбционном анализе до последнего времени чаще всего использовали пламя (2), представляющее собой низкотемпературную плазму (пламя горючих газов в смеси с окислителями). При этом необходимыми условиями являлись прозрачность пламени во всем спектральном интервале; слабое собственное излучение пламени; большая эффективность атомизации элемента в пламени. Наибольшее распространение получили пламя "воздух - ацетилен" (Тm ах = 2300 °С) и "оксид азота N2 0 - ацетилен" (Тm ах = 2950 °С). Первое обеспечивает высокую эффективность атомизации более 30 элементов, в том числе щелочных и щелочно-земельных; во втором возможно определении почти всех элементов периодической системы, но оно имеет интенсивное собственное излучение в некоторых участках спектра, для устранения которого к пробе добавляется легко ионизирующий металл. В атомно-абсорбционной спектроскопии пламя формируется в горелке с длинной щелью, чтобы увеличить длину поглощающего света.
Альтернативой пламени служит электротермический атомизатор (ЭТА). В таком атомизаторе используют электрический нагрев тугоплавкого материала, на который наносят пробу. Таким образом, здесь реализуется нестационарное образование свободных атомов. Значительное преимущество ЭТА по сравнению с пламенем заключается в увеличении времени пребывания свободных атомов.
Для выделения узкого участка спектра служит мопохроматизатор (3) - устройство получения света с заданной длиной волны. Его основные детали - щели, линзы, зеркала и диспергирующие элементы, которые разлагают излучение в спектр - дают раздельное изображение спектральных линий (призмы из стекла и кварца и дифракционные решетки). Призмы из стекла используют в видимом и инфракрасном участке спектра, кварцевые призмы - в УФ области спектра, дифракционные решетки - в области спектра от 200 до 1000 им. Осветительная система атомно-абсорбционного спектрометра фокусирует свет источника на входную щель монохроматора.
Детектор - приемник света (4) - преобразует падающую на него световую энергию в электрический сигнал. В атомно-абсорбционном анализе для этой цели всегда используют фотоэлектронные умножители. В них поглощение света либо приводит к отрыву электрона с облучаемой поверхности, либо к увеличению электрической проводимости под действием света.
Спектрометр с электротермическим атомизатором состоит из источника излучения (1), оптической системы (2), электротермического атомизатора, включающего графитовую трубчатую печь (3) и электромагнит (4), монохроматора (5), фотоэлектрического преобразователя (6) и персонального компьютера (7).

Рис.2. Схема атомно-абсорбционного спектрометра с электротермической атомизацией пробы
Свет от источника резонансного излучения (1) с помощью двухлинзовой оптической системы (2) пропускается через графитовую печь (3), которая расположена в воздушном зазоре электромагнита (4), питающегося однофазовым сетевым напряжением.
При испарении пробы в аналитической ячейке кроме атомов определяемого элемента может присутствовать фон (частицы и молекулы), поглощение света которым приводит к появлению систематической погрешности атомно-абсорбционных измерений. Для автоматической коррекции фонового поглощения в спектрофотометре использован обратный эффект Зеемана - графитовая печь помещена в продольное переменное магнитное поле.
Электрический ток подается через массивные графитовые токоподводящие контакты на тонкостенную, полую внутри графитовую трубку и нагревает ее. Напряжение регулируется в интервале от 0 до 10 В, при этом сила тока, проходящего через графитовую трубку меняется от 0 до 400 А, а температура печи - от комнатной до 3100 0 С.
Аликвота анализируемой пробы - до 10 мкл (в виде раствора) вводится в отверстие графитовой трубки ручным дозатором (микропипеткой). За счет мощного дугового разряда проба мгновенно испаряется. Через каналы в графитовых электродах вокруг трубки циркулирует инертный газ, который через отверстие для пробы входит внутрь трубки, а через ее открытие концы выходит в атмосферу. Газ предохраняет атомизатор от воздействия атмосферного кислорода и способствует удалению из печи атомизированной пробы. Графитовая кювета располагается внутри охлаждаемого водой кожуха. Температура графитовой печи регулируется устройством с программным управлением (ЭВМ), позволяя разделить во времени процессы высушивания, сгорания, атомизации пробы и очистки печи от продуктов сгорания.
На стадии атомизации в печи возникает атомный пар, содержащий атомы определяемого компонента и фоновые образования. В магнитном поле происходит расщепление линии поглощения определяемых атомов. Недостатком электротермического метода атомизации пробы является меньшее количество определяемых элементов, меньшая воспроизводимость результатов анализа и возможность воздействия материала печи на условия атомизации (например, путем образования карбидов).
Интенсивность света, прошедшего через аналитическую ячейку, определяется выражением:
I= I0 ехр (- kν • с - k ф ) • l,
где I 0 - интенсивность света на входе аналитической ячейки; l - длина аналитической ячейки; с - концентрация определяемых атомов в ячейке; kν - коэффициент абсорбции;
k ф - коэффициент фонового поглощения, независящий от магнитной индукции.
Свет, прошедший через графитовую печь, с помощью линз фокусируется на входной щели монохроматора (5), который выделяет спектральный интервал, содержащий используемую резонансную линию определяемого элемента. Пройдя через монохроматор, свет поступает на фотоэлектрический преобразователь (6), напряжение, на выходе которого пропорционально интенсивности излучения. Этот сигнал формируется в цифровые сигналы, которые передаются компьютеру (7), где вычисляется выходной сигнал истинной абсорбционности, равной разности сигналов суммарной (lglmax ) (в случае максимального расщепления линии поглощения) и фоновой абсорбционности (lgI 0 ) (отсутствие расщепления):
A = lgImax - lgI0
Сигнал абсорбционности не зависит ни от интенсивности источника излучения, ни от коэффициента фонового поглощения света, а зависит только от концентрации определяемого элемента в аналитической ячейке, прямо пропорциональной концентрации элемента в анализируемой пробе.
В процессе атомизации сигнал атомной абсорбционности нарастает от нуля до амплитудного значения (пик сигнала), а затем опять падает до нуля. Пик сигнала зависит от концентрации определяемого элемента и служит основным информационным параметром, по которому производится вычисление концентрации элемента в пробе по градуировочной зависимости, в качестве которого используется квадратичный полином А = а + bС + сС2 . Коэффициенты полинома вычисляются в процессе градуировки. Статистическая обработка результатов измерений заключается в вычислении доверительных интервалов концентрации с учетом случайной составляющей погрешности измерения абсорбционности и расчета градуировочных коэффициентов.
Химические помехи
Как пламенная, так и электротермическая атомно-абсорбциониая спектроскопия могут быть чувствительны к влиянию основы пробы, вызывающих изменения при образовании свободных атомов. В случае пламени ограниченная температура не обеспечивает полной диссоциации и атомизации термически устойчивого соединения в газовой фазе. Хорошо известным примером является влияние фосфата на кальций, что приводит к образованию устойчивых фосфатов кальция.
Спектральные помехи
В атомно-абсорбционной спектроскопии возможность спектральных помех от спектральной линии другого элемента, попадающей в спектральную полосу пропускания диспергирующей системы, крайне мала. Большие трудности создает наличие неспецифического поглощения света компонентами основы. Это может приводить к значительному увеличению фона, и, следовательно, к увеличению сигнала. Чтобы компенсировать данное увеличение фона, необходимо вычитать фон. Наиболее широко используется метод дейтериевой лампы и метод, основанный па эффекте Зеемана (расщепление спектральных линий в магнитном поле).
Преимущества атомно-абсорбционного метода можно сформулировать следующим образом: метод обладает высокой селективностью, имеет низкие пределы обнаружения, позволяет определять несколько элементов из одного раствора, подготовка проб к измерениям простая, процедура анализа автоматизирована.
Глава II.
1. Карта-схема расположения участков пробоотбора
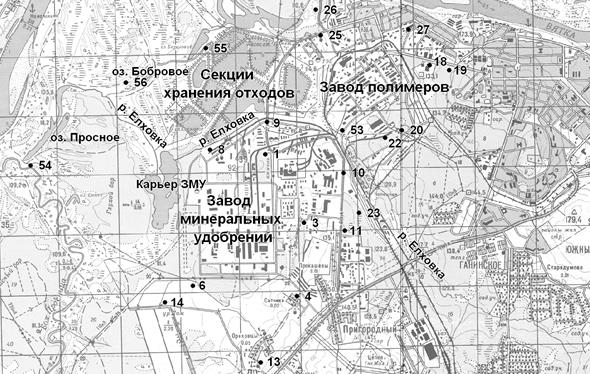
Пробы снега отбирали в марте 2011 г. на площадках с ненарушенным снеговым покровом в зоне влияния КЧХК,которые располагались по восьми румбам от источника загрязнения – завода минеральных удобрений (ЗМУ) (см. карту).
С каждого участка на всю высоту снегового покрова отбирали одну смешанную пробу, состоящую из трех–четырех точечных проб. Фоновый участок находился в поле рядом с лесным массивом с. Тохтино Орловского района Кировской области.
2. Методика проведения химического анализа и обработка результатов
2.1. Атомно-абсорбционная спектрометрия
Метод заключается в обработке измельченной сухой пробы почвы соответствующими реагентами и последующим атомно-абсорбционным определением металлов в пламени пропан-воздух или ацетилен-воздух.
При определении содержания металлов, близких к пределу обнаружения их можно сконцентрировать непосредственно в процессе измерения. Концентрирование производится автоматически путем накопления в течение 1 - 2 минут определяемого металла на микроколонке с хелатным сорбентом, через который пропускается раствор пробы, затем накопленный металл в течение 10-15 секунд вводится в пламя атомизатора.
В результате достигается снижение предела обнаружения в 10-25 раз.
Проведение анализа
1. Подготовка проб.
Химическое разложение проб почв при валовом определении тяжелых металлов.
10 г воздушно-сухой почвы, измельченной и пропущенной через сито с отверстием 2 мм, взвешивают на технических весах, помещают навеску в химический стакан или концентрическую колбу вместимостью 200 - 250 см3 и приливают 50 см3 HN03 (1:1). При содержании в почве свыше 5% гумуса рекомендуется предварительное сухое озоление пробы при 575 0 С (по Тюрину).
Стакан закрывают часовым стаканом и помещают на закрытую электроплитку, доводят до кипения и кипятят на медленном огне 10 мин, затем к пробе по каплям приливают 10 см3 концентрированной перекиси водорода при перемешивании и вновь помещают на электроплитку, доводят до кипения и кипятят еще 10 мин.
После охлаждения до комнатной температуры суспензию отфильтровывают через складчатый фильтр «синяя лента» в мерную колбу вместимостью 100 см3 , фильтр с осадком помещают в стакан, в котором остался остаток почвы, приливают в стакан 40 см3 1М азотной кислоты и помещают его на плитку, нагревают и кипятят еще 30 мин.
После охлаждения до комнатной температуры жидкость в стакане отфильтровывают в ту же мерную колбу. Осадок на фильтре промывают горячей азотной кислотой с концентрацией 1 моль/дм3 и после охлаждения доводят объем фильтра в мерной колбе до метки бидистиллированной водой.
Одновременно проводят «холостой» анализ, включая все его стадии, кроме взятия пробы почвы.
Экстракция подвижных форм тяжелых металлов из почв с помощью кислот.
Подвижные кислоторастворимые формы, металлов (Си, Zn, Ni, Со, Cd,Pb) определяют в вытяжках 1М HNO3 или 1М НС1.
В последние годы эти экстрагенты успешно используют для анализа почв, подверженных техногенным воздействиям. Из сильно загрязненных почв 1М HNO3 извлекают 90 - 95% тяжелых металлов от их валового содержания. Отношение почвы к раствору 1:10, для торфяных почв 1:20.
Пробы почвы массой 5г. (для торфяных почв 2,5г.) взвешивают с точностью ±0,1 г. и помещают коническую колбу вместимостью 200 - 300 см3 , к пробе добавляют 50 см3 1М HNO3 (для извлечения РЬ можно использовать 1М НС1). Навеску почвы необходимо увеличить до 10г. при определении тяжелых металлов на фоновом уровне. При этом соотношение почвы и раствора остается неизменным.
Взбалтывают суспензию на ротаторе в течение 1 часа или после 3-минутного встряхивания настаивают в течение суток. Колбу закрывают пробкой (если резиновая, то ее необходимо завернуть полиэтиленовой пленкой.).
Вытяжку фильтруют через сухой складчатый фильтр «белая лента», предварительно промытый 1М HNO3 . Перед фильтрованием вытяжка перемешивается, и переносится на фильтр по возможности полностью. В фильтрате определяют тяжелые металлы на атомно-абсорбционном спектрофотометре в пламени ацетилен-воздух. Если фильтраты мутные, их возвращают на фильтры. Одновременно проводят холостой анализ, включая все стадии его определения, кроме взятия проб.
Извлечение подвижных форм тяжелых металлов ацетатно-аммонийным буферным раствором с рН = 4,8.
Подвижные формы соединении элементов в почвах извлекают ацетатно-аммонийным буфером раствором с рН = 4,8 (ААБ). Этот экстрагент принят агрохимической службой для извлечения доступных растениям микроэлементов и служит для оценки обеспеченности почв этими элементами.
Отношение почвы к раствору 1:10, время воздействия 1 час при взбалтывании на ротаторе или настаивании в течение суток. Метод пригоден для некарбонатных и карбонатных почв. При анализе торфяных почв отношение почв к раствору должно быть увеличено до 1:20.
Пробу почвы массой 10г. помещают в коническую колбу вместимостью 250 см3 , приливают 50 см3 ацетатно-аммонийного буфера.
Суспензию взбалтывают 1 час и настаивают в течение суток. Суспензии карбонатных почв, не закрывая емкости, периодически взбалтывают от руки до прекращения выделения углекислого газа. Вытяжки фильтруют через сухой складчатый фильтр «белая лента», по возможности не перенося почву на фильтр. К оставшейся в колбе почве приливают 50 см3 ацетатно-аммонийного буфера и экстрагирование повторяют, повторное фильтрование производят в ту же колбу, перенося на фильтр максимальное количество почвы.
Одновременно проводят холостой анализ, включая все его стадии, кроме взятия проб.
При измерении концентрации металлов в экстрактах почв близких к пределу обнаружения целесообразно воспользоваться блоком проточного концентрировании БК-1, в котором происходит селективное концентрирование металлов в колонке с сорбентом и последующее элюирование концентрата 2М HNO3 в детектор без размывания зоны концентрата с последующим атомно-абсорбционным определением содержания металлов в концентрате.
Этот метод пригоден для некарбонатных и карбонатных почв. При анализе торфяных почв отношение почвы к раствору должно быть увеличено до 1:20.
2. Подготовка анализатора к работе и выбор условий измерения.
Подготовка анализа к работе, его включение и выведение на рабочий режим осуществляется в соответствии с руководством по эксплуатации, прилагаемой к прибору.
Выбор условий измерения определяют из справочника данных по определяемому элементу заложенных в память компьютера.
При анализе почв необходимо работать с использованием коррекции фона (двухимпульсный режим), из-за наличия неселективной абсорбции.
3. Проведение измерений.
В режиме «Градуировка» (анализатор работает совместно с компьютером) измеряют абсорбцию растворов сравнения в порядке возрастания концентрации.
После построения градировочного графика, в режиме «Анализ» проводят измерения концентрации металла в экстрактах почв. После измерения концентрации каждой десятой пробы измеряют один из растворов сравнения по которому проводилась градуировка. Включают режим «Рекалибровка». При отклонении градуировочного коэффициента более, чем на 10% от установленного ранее, заменяют старую градуировку новой и повторяют замер этих десяти проб. Следует, однако, выяснить причину изменения градуировки (засорился распылитель, сбилась линия, изменился расход газа) и по возможности устранить ее.
Расчетное содержание металла считывают с экрана монитора или распечатывают на принтере.
Проведение измерений концентрации каждого раствора проводят не
менее двух раз.
Обработка результатов измерения
Содержание металлов в исследуемых пробах почв рассчитывают по формуле:
X= ![]() ;
;
где X - массовая доля определяемого металла в воздушно-сухой пробе, мг/кг.
![]() - концентрация металла в исследуемой кислотной (буферной) вытяжке почвы, мг/дм3
.
- концентрация металла в исследуемой кислотной (буферной) вытяжке почвы, мг/дм3
.
![]() - концентрация металла в холостой пробе, мг/дм3
.
- концентрация металла в холостой пробе, мг/дм3
.
V - объем исследуемого раствора, см3 , m - навеска, г.
За результат измерения массовой доли металла принимают среднеарифметическое значение mх двух результатов параллельных определений m1 и m2 .
2.2. Вольтамперометрия
1. Подготовка проб.
При подготовке к выполнению измерений должны быть проведены следующие работы: приготовление анализируемого раствора пробы (при необходимости - разрушение органических веществ минерализацией до влажных солей или фотохимическое окисление пробы; удаление ионов Сu (II) перед определением массовой концентрации ионов цинка), подготовка электродов, анализатора "Экотест-ВА" и сборка электрохимической ячейки.
Все пробы консервируют азотной кислотой из расчета 5 см3 кислоты на 1 дм3 пробы до значения рН пробы 1,5-2,5, контролируя рН с помощью рН-метра-иономера "Эксперт-001". В случае щелочных природных, морских, минеральных и сточных вод дополнительно добавляют концентрированную азотную кислоту и доводят рН раствора до значения 1,5-2,5. Подкисленные пробы хранят при температуре 3 - 4 °С не более месяца.
2. Приготовление анализируемого раствора пробы воды.
При измерении концентрации суммы форм ионов металлов подкисленную пробу переносят в термостойкий стакан, нагревают на плитке в течение 30-40 мин при температуре 80-90 °С, охлаждают до температуры 20-30 °С и фильтруют через обеззоленный фильтр Фильтрат используют для приготовления анализируемого раствора пробы.
100 см3 пробы переносят в фарфоровую чашку. Добавляют 2 см3 концентрированной азотной кислоты и упаривают раствор до влажных солей на электроплитке со слабым нагревом. Минерализация считается законченной, если остаток осветлился. К остатку добавляют 1 см3 1 М соляной кислоты и упаривают раствор досуха на водяной бане. После окончания минерализации переходят к приготовлению анализируемого раствора пробы.
Если остаток остается темный, кислотную обработку повторяют до его осветления (3-5 раз). Если остаток не осветляется после кислотной обработки, пробу упаривают досуха, помещают в муфельную печь и прокаливают при температуре не выше 400 °С в течение 30 - 50 мин. Чашку с золой вынимают из муфельной печи, охлаждают до комнатной температуры. Минерализация считается законченной, когда зола станет светлого цвета. Золу смачивают 1 см3 1 М соляной кислоты и досуха выпаривают на водяной бане. После окончания минерализации переходят к приготовлению анализируемого раствора пробы.
Для приготовления раствора чашку с осадком охлаждают до комнатной температуры. К осадку добавляют 1 см3 1 М соляной кислоты и 10 см3 разбавленного фонового раствора, тщательно перемешивают. Раствор фильтруют через обеззоленный фильтр в мерную колбу вместимостью 100 см . Смывают чашку и фильтр 15-20 см3 разбавленного фонового раствора и вносят смыв в колбу. Доводят объем раствора до 100 см3 разбавленным фоновым раствором.
25 см3 анализируемого раствора пробы вносят в электрохимическую ячейку и проводят анализ.
При приготовлении анализируемого раствора пробы для измерения массовой концентрации ионов цинка пробу анализируемого раствора объемом 30-35 см3 пропускают через концентрирующий патрон. 25 см3 очищенного от ионов меди анализируемого раствора пробы вносят в электрохимическую ячейку для анализа.
3. Подготовка электродов.
Рабочий углеситалловый электрод. Перед работой торец электрода протирают фильтровальной бумагой, смоченной в этиловом спирте. Перед измерением три раза ополаскивают бидистиллированной водой и просушивают фильтровальной бумагой.
Электрод сравнения заполняют насыщенным раствором хлористого калия, таким образом, чтобы не было пузырьков воздуха. При первом заполнении электрод выдерживают не менее 24 часов. Перед измерением электрод три раза ополаскивают бидистиллированной водой и просушивают фильтровальной бумагой. Между измерениями электрод хранят в насыщенном растворе хлористого калия.
Вспомогательный электрод - стеклоуглеродный стаканчик датчика "Модуль ЕМ-04" перед измерением промывают раствором 1 М соляной кислоты, три раза ополаскивают бидистиллированной водой и просушивают фильтровальной бумагой.
4. Подготовка к работе анализатора.
Анализатор "Экотест-ВА" подключают к персональному компьютеру согласно "Руководству по эксплуатации" на анализатор. Установление программного обеспечения и запуск программы анализатора проводят в соответствии с "Руководством по эксплуатации" на анализатор.
5. Сборка электрохимической ячейки.
Собирают электрохимическую ячейку в соответствии с описанием на датчик "Модуль ЕМ-04". Соединяют анализатор "Экотест-ВА" с датчиком "Модуль ЕМ-04" согласно "Руководству по эксплуатации" на анализатор.
При подключении к электрической сети на передней панели анализатора засветится сигнальный светодиод, а на цифровом индикаторе панели управления датчика "Модуль ЕМ-04" загорится число 1000, предусмотренная методикой определения скорость вращения мешалки (число оборотов в мин).
6. Выполнение измерений.
Одновременно анализируют не менее двух параллельных проб воды.
6.1. Регистрация вольтамперограмм анализируемого раствора пробы по и раствора пробы с добавками стандартных растворов Zn(II), Cd (II) и Pb (II) после накопления при потенциале-1300 мВ.
6.2. 25 см3 контрольной пробы, подготовленной к измерению по п.1.8.2.5., помещают в электрохимическую ячейку. Запускают программу «ВА-95». Работу с программой «ВА-95» проводят согласно «Руководству оператора». Параметры электрохимического измерения устанавливают согласно таблице представленной ниже.
| Наименование параметра | Единицы измерения | Величина параметра |
| Скорость развертки потенциала | мВ/с В/с | от 50 до 100 |
| Начало развертки потенциала | мВ | минус 950 для Сu, минус 1300 для Zn, Cd, Pb |
| Конец развертки потенциала | мВ | 200 |
| Потенциал накопления | мВ | минус 950 при определении Сu, минус 1300 при определении Zn, Cd, Pb |
| Продолжительность накопления | с | 300 |
| Мешалка | Вкл | |
| Продолжительность успокоения раствора в ячейке | с | 10 |
| Потенциал очистки электрода | мВ | 100 |
| Продолжительность очистки электрода | с | равна продолжительности накопления |
| Шкала измерения анодного тока | мкА | диапазон 0/20; 0/200 |
7. Обработка вольтамперограммы контрольной пробы.
Измерение площади или высоты аналитического сигнала ионов металла производят в соответствии с «Руководству оператора».
Измерение вольтамперограммы и ее обработку повторяют не менее 3-х раз до сходимости результатов, т.е. до момента, когда разность высот пиков определяемых ионов металла в двух последних вольтамперограммах не превышает 5 % от высоты пика.
Регистрация и обработка вольтамперограммы анализируемого раствора пробы .
При регистрации вольтамперограммы анализируемого раствора пробы используют подготовленные растворы. 25 см3 вносят в электрохимическую ячейку. Активизируют окно "Параметры измерения" и вносят необходимые изменения в параметры продолжительности накопления, очистки электрода и чувствительности (шкала токов) анализатора, либо загружают стандартную методику анализа, заложенную в программное обеспечение «ВА-95». Активируют команду "Старт" для начала измерений.
Измерение вольтамперограмм проводят не менее трех раз до сходимости их характеристик. При необходимости вносят изменения в параметры измерений и повторяют запись вольтамперограммы пробы.
Регистрация и обработка вольтамперограммы анализируемого раствора пробы с добавками стандартных растворов ионов металлов.
В ячейку с анализируемым раствором пробы вносят пипеткой добавки стандартных растворов ионов металлов. Рекомендуемый объем добавки такой, чтобы высота пика ионов металла после внесения добавки увеличивалась бы в 1,5-2 раза. Общий объем добавленных стандартных растворов не должен превышать 10% от исходного объема раствора в ячейке.
Обработку вольтамперограммы проводят не менее 3-х раз до сходимости результатов аналогично обработке вольтамперограммы анализируемого раствора пробы.
8. Обработка результатов измерений.
Концентрацию ионов металла в анализируемом растворе находят по формуле:

где Сm – концентрация ионов металла в анализируемом растворе пробы, мкг/л,
Sx – площадь пика ионов металла в анализируемом растворе пробы,
Sф- площадь пика ионов металла в растворе контрольной пробы,
S – площадь пика ионов металла в анализируемом растворе пробы с добавкой стандартного раствора ионов металла,
V – объём раствора в ячейке до внесения добавки, мл.,
Vд – объём добавки стандартного раствора ионов металла, мл.,
Cд – концентрация добавленного стандартного раствора ионов металла, мкг/л.
Расчёт проводится в программномобеспечению анализатора.
9. Оформление результатов измерения.
За результат анализа С принимают среднее арифметическое результатов параллельных определений С1 и С2 (С = (С1 +С2 /2),расхождение между которыми не превосходит значений норматива оперативного контроля r.
Результат количественного анализа в документах, предусматривающих его использование, представляют в виде:
С + Δ, мкг/дм3 , где С - результат анализа (мкг/дм3 ), Δ - показатель точности (мкг/дм3 ) (по таблице ниже)
или С - результат анализа (мкг/дм3 ), δ(%) - относительный показатель точности, где δ(%)=100Δ/С.
Результат измерений должен оканчиваться тем же десятичным разрядом, что и погрешность.
| Диапазон измерений в анализируемом растворе пробы, мкг/дм3 | Повторяемость, % | Воспроизводимость, % | Граница систематической погрешности, % | Граница точности, % | |||
| Cd | |||||||
| от 0,5 до 2,0 | 11 | 14 | 12 | 28 | |||
| от 2,0 до 5.0 | 8 | 11 | 10 | 22 | |||
| от 5,0 до 500 вкл | 5 | 7 | 6 | 14 | |||
| Рb | |||||||
| от 0,5 до 1,0 | 13 | 17 | 15 | 34 | |||
| от 1,0 до 100 | 10 | 13 | 11 | 25 | |||
| от 100 до 500 вкл | 6 | 9 | 8 | 17 | |||
| Сu | |||||||
| от 1,0 до 5,0 | 20 | 22 | 14 | 43 | |||
| от 5,0 до 10 | 16 | 10 | 17 | 37 | |||
| от 10 до 50 | II | 14 | 12 | 28 | |||
| от 50 до 500 вкл | 10 | 12 | 11 | 24 | |||
| Zn | |||||||
| от 1,0 до 5,0 | 12 | 16 | 14 | 32 | |||
| от 5,0 до 100 | 8 | 11 | 10 | 22 | |||
| от 100 до 500 вкл. | 5 | 6 | 5 | 12 | |||
Глава III.
1. Результаты.
Результаты по иону Cu
| № участка | Содержание мкг/л | № участка | Содержание мкг/л |
| 55 | 0,8 | 55 | 9 |
| 56 | 9,8 | 56 | 10 |
| 54 | 1,2 | 54 | 7,5 |
| 26 | 7,9 | 26 | 9 |
| 14 | 9,2 | 14 | 5,5 |
| 23 | 7,8 | 23 | 10 |
| 1 | 0,8 | 1 | 9 |
| 4 | 2,5 | 4 | 8 |
| 6 | 0,5 | 6 | 7 |
| 53 | 4,7 | 53 | 9 |
| 8 | 8,9 | 8 | 9,5 |
| 25 | 8,8 | 25 | 8,5 |
| 22 | 8,9 | 22 | 7,5 |
| 18 | 0,8 | 18 | 9,5 |
| 27 | 4,4 | 27 | 5 |
| 19 | 0,9 | 19 | 5,5 |
| 13 | 0,5 | 13 | 8 |
| 3 | 2,2 | 3 | 7,5 |
| 20 | 2,8 | 20 | 7 |
| 10 | 7,8 | 10 | 9 |
| 9 | 1 | 9 | 5 |
| 11 | 2,5 | 11 | 10 |
| ВАМ | ААС | ||
График зависимости ААС и ВАМ по иону Cu (II), где цвентыми линиями отмечена граница фона, а цвет линии характеризует соответствующий метод анализа.

Результаты по иону Pb
| № участка | Содержание мкг/л | № участка | Содержание мкг/л |
| 55 | 1,7 | 55 | 3 |
| 56 | 5,4 | 56 | 6 |
| 54 | 2,2 | 54 | 5 |
| 26 | 1,4 | 26 | 9 |
| 14 | 2,6 | 14 | не обнаружено |
| 23 | 10,2 | 23 | 10 |
| 1 | 3,2 | 1 | 15 |
| 4 | 1,8 | 4 | 9,5 |
| 6 | 1,9 | 6 | не обнаружено |
| 53 | 9,3 | 53 | 4 |
| 8 | не обнаружено | 8 | 9 |
| 25 | 3,9 | 25 | 7,2 |
| 22 | 3 | 22 | 8 |
| 18 | 2,6 | 18 | 3,5 |
| 27 | 0,9 | 27 | 2,3 |
| 19 | 4,8 | 19 | 3,7 |
| 13 | 0,6 | 13 | 0,8 |
| 3 | 6,1 | 3 | 4,4 |
| 20 | 29,4 | 20 | 30 |
| 10 | 8,7 | 10 | 4,5 |
| 9 | 6,1 | 9 | 2 |
| 11 | 2,3 | 11 | 1 |
| ВАМ | ААС | ||
График зависимости ААС и ВАМ по иону Pb(II) , где цвентыми линиями отмечена граница фона, а цвет линии характеризует соответствующий метод анализа.

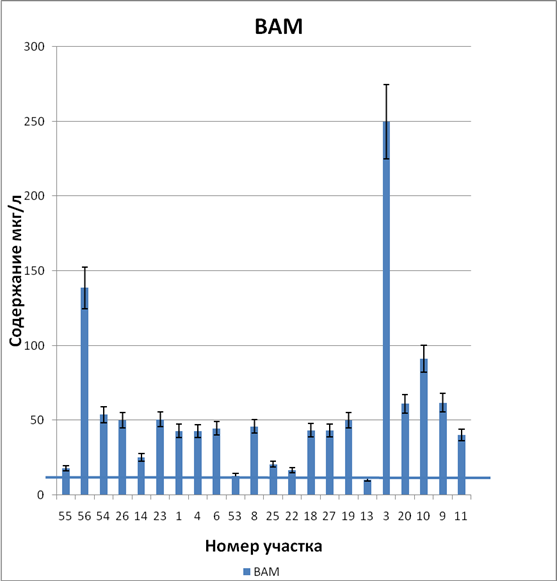
Результаты по иону Zn
| № участка | Содержание мкг/л |
| 55 | 18 |
| 56 | 138,7 |
| 54 | 53,8 |
| 26 | 50 |
| 14 | 25,2 |
| 23 | 50,7 |
| 1 | 43 |
| 4 | 42,9 |
| 6 | 44,7 |
| 53 | 13 |
| 8 | 46 |
| 25 | 20,7 |
| 22 | 16,6 |
| 18 | 43,4 |
| 27 | 43,2 |
| 19 | 50 |
| 13 | 10,5 |
| 3 | 249,7 |
| 20 | 61,1 |
| 10 | 91,3 |
| 9 | 61,9 |
| 11 | 40,2 |
| ВАМ | |
График зависимости ВАМ по иону Zn (II) , где синей линией отмечена граница фона.
2. Заключение
1. Установлено, что на большинстве участков вблизи КЧХК содержание тяжелых металлов (цинка, меди, свинца) в снеге превышает значения фона. Однако абсолютные значения концентрации данных металлов в снеге не высоки.
2. Высокое содержание меди и цинка в снеге, в 1,5-5 раз превышающее фон, установлено на участке 56, который находится вблизи шламонакопителя отходов Кирово-Чепецкого химического комбината. В 3 раза превышала фоновое значение концентрация свинца Pb в снеге участка 20, расположенного рядом с автодорогами. Максимальное превышение фона (в 15 раз) установлено для ионов цинка на участке 3, что может связано с близостью железной дороги.
3. Различие данных, полученных методами инверсионной вольтамперометрии и атомно-абсорбционной спектрометрии, обусловлено различиями пределов обнаружения. Нижний предел обнаружения тяжелых металлов ВАМ (0,5-1 мкг/л) на порядок ниже по сравнению с ААС (5-10 мкг/л). В связи с этим для получения достоверных результатов по содержанию тяжелых металлов в снеге методом ААС, необходимо проводить предварительное концентрирование пробы.