| Скачать .docx |
Дипломная работа: Создание новых лекарственных веществ
Введение
Несмотря на достижения современной анестезии, продолжаются поиски менее опасных средств для наркоза, разработка различных вариантов многокомпонентного избирательного наркоза, позволяющего значительно уменьшить их токсичность и побочные отрицательные влияния.
Создание новых лекарственных веществ включает 6 стадий:
1. Создание лекарственного вещества с помощью компьютерного моделирования.
2. Лабораторный синтез.
3. Биоскрининг и доклинические испытания.
4. Клинические испытания.
5. Промышленное производство.
6. Продажа.
В последнее время компьютерное моделирование все более уверенно входит в практику технологии создания новых синтетических лекарственных веществ [1]. Предварительно проведенный компьютерный скрининг экономит время, материалы и силы при аналоговом поиске лекарственных препаратов. В качестве объекта исследования выбран местноанестезирующий препарат дикаин, который имеет более высокий уровень токсичности в ряду своих аналогов, но при этом не заменим в глазной и оториноларингологической практике. Для снижения и сохранения или усиления местноанестезирующего эффекта разрабатываются композиционные составы, дополнительно содержащие противогистаминные средства, содержащих аминоблокаторы, адреналин.
Дикаин относится к классу сложных эфиров п -аминобензойной кислоты (β-диметиламиноэтиловый эфир п -бутиламинобензойной кислоты гидрохлорид) [2]. Расстояние C-N в 2-аминоэтанольной группе определяет двухточечный контакт молекулы дикаина с рецептором через диполь-дипольное и ионное взаимодействие.
В основу модифицирования молекулы дикаина для создания новых анестетиков нами положен принцип введения химических группировок и фрагментов в существующий анестезиофор, которые усиливают взаимодействие вещества с биорецептором, снижают токсичность и дают метаболиты с положительным фармакодействием.
Исходя из этого нами предложены следующие варианты новых молекулярных структур:
1. В бензельное кольцо введена “облагораживающая” карбоксильная группа, диметиламиногруппа замещена на более фармакоактивную диэтиламиногруппу.
2. Алифатический н -бутильный радикал замещен на адреналиновый фрагмент.
3. Ароматическая основа п -аминобензойной кислоты замещена на никотиновую кислоту.
4. Бензольное кольцо замещено на пиперидиновое, характерное для эффективного анестетика промедол.
В работе выполнено компьютерное моделирование всех указанных структур с применением программы HyperChem. На последующих этапах компьютерного конструирования исследована биологическая активность новых анестетиков с применением программы PASS.
1. Обзор литературы
1.1 Лекарственные средства
Несмотря на огромный арсенал имеющихся лекарств, проблема изыскания новых высокоэффективных лекарственных средств остается актуальной. Это обусловлено отсутствием или недостаточной эффективностью лекарств для лечения некоторых заболеваний; наличие побочного действия некоторых лекарственных препаратов; ограничениями срока годности лекарственных препаратов; огромными сроками годности лекарственных препаратов или их лекарственных форм.
Создание каждого нового оригинального лекарственного вещества является результатом развития фундаментальных знаний и достижений медицинских, биологических, химических и других наук, проведения напряженных экспериментальных исследований, вложения крупных материальных затрат. Успехи современной фармакотерапии явились следствием глубоких теоретических исследований первичных механизмов гомеостаза, молекулярных основ патологических процессов, открытия и изучения физиологически активных соединений (гормоны, медиаторы, простагландины и др.) [3]. Получению новых химиотерапевтических средств способствовали достижения в изучении первичных механизмов инфекционных процессов и биохимии микроорганизмов.
Лекарственное средство – однокомпонентный или комплексный состав, обладающий профилактической и лечебной эффективностью. Лекарственное вещество – индивидуальное химическое соединение, используемое в качестве лекарственного средства [4].
Лекарственная форма – физическое состояние лекарственного средства, удобное для применения [4].
Лекарственный препарат – дозированное лекарственное средство в адекватной для индивидуального применения лекарственной форме и оптимальным оформлением с приложением аннотации о его свойствах и использовании [4].
В настоящее время каждое потенциальное лекарственное вещество проходит 3 стадии изучения: фармацевтическую, фармакокинетическую и фармакодинамическую.
На фармацевтической стадии устанавливают наличие полезного действия лекарственного вещества, после чего оно подвергается доклиническому изучению других показателей. Прежде всего определяется острая токсичность, т.е. смертельная доза для 50% опытных животных. Затем выясняется субхроническая токсичность в условиях длительного (несколько месяцев) введения лекарственного вещества в терапевтических дозах. При этом наблюдают возможные побочные эффекты и патологические изменения всех систем организма: тератогенность, влияние на репродуктивность и иммунную систему, эмбриотоксичность, мутагенность, канцерогенность, аллергенность и другие вредные побочные действия. После этого этапа лекарственное средство может быть допущено к клиническим испытаниям.
На второй стадии - фармакокинетической - изучают судьбу лекарственного вещества в организме: пути его введения и всасывания, распределение в биожидкостях, проникновение через защитные барьеры, доступ к органу-мишени, пути и скорость биотрансформации пути выведения из организма (с мочой, калом, потом и дыханием).
На третьей - фармакодинамической - стадии изучаются проблемы распознавания лекарственного вещества (или его метаболитов) мишенями и их последующего взаимодействия. Мишенями могут служить органы, ткани, клетки, клеточные мембраны, ферменты, нуклеиновые кислоты, регуляторные молекулы (гормоны, витамины, нейромедиаторы и т.д.), а также биорецепторы. Рассматриваются вопросы структурной и стереоспецифичной комплементарности взаимодействующих структур, функционального и химического соответствия лекарственного вещества или метаболита его рецептору. Взаимодействие между лекарственным веществом и рецептором или акцептором, приводящее к активации (стимулированию) или дезактивации (ингибированию) биомишени и сопровождающееся ответом организма в целом, в основном обеспечивается за счет слабых связей – водородных, электростатических, ван-дер-ваальсовых, гидрофобных [5].
1.2 Создание и исследование новых лекарственных средств. Основное направление поиска
Создание новых лекарственных веществ оказалось возможным на основе достижений в области органической и фармацевтической химии, использования физико-химических методов, проведения технологических, биотехнологических и других исследований синтетических и природных соединений.
Общепринятым фундаментом создания теории целенаправленных поисков тех или иных групп лекарственных препаратов является установление связей между фармакологическим действием и физическими особенностями [3].
В настоящее время поиск новых лекарственных средств ведется по следующим основным направлениям.
1. Эмпирическое изучение того или иного вида фармакологической активности различных веществ, полученных химическим путем. В основе этого изучения лежит метод «проб и ошибок», при котором фармакологи берут существующие вещества и определяют с помощью набора фармакологических методик их принадлежность к той или иной фармакологической группе. Затем среди них отбирают наиболее активные вещества и устанавливают степень их фармакологической активности и токсичности по сравнению с существующими лекарственными средствами, которые используются в качестве стандарта.
2. Второе направление состоит в отборе соединений с одним определенным видом фармакологической активности. Это направление получило название направленного изыскания лекарственных средств.
Преимущество этой системы состоит в более быстром отборе фармакологически активных веществ, а недостатком является отсутствие выявления других, может быть весьма ценных видов фармакологической активности.
3. Следующее направление поиска – модификация структур существующих лекарственных средств. Этот путь поиска новых лекарственных средств является теперь весьма распространенным. Химики-синтетики заменяют в существующем соединении один радикал другим, вводят в состав исходной молекулы другие химические элементы или производят иные модификации. Этот путь позволяет увеличить активность лекарственного препарата, сделать его действие более избирательным, а также уменьшить нежелательные стороны действия и его токсичность [4].
Целенаправленный синтез лекарственных веществ означает поиск веществ с заранее заданными фармакологическими свойствами. Синтез новых структур с предполагаемой активностью чаще всего проводится в том классе химических соединений, где уже найдены вещества, обладающие определенной направленностью действия на данный орган или ткань.
Для основного скелета искомого вещества могут быть выбраны также те классы химических соединений, к которым относятся естественные вещества, участвующие в осуществлении функций организма. Целенаправленный синтез фармакологических веществ труднее вести в новых химических классах соединений ввиду отсутствия необходимых первоначальных сведений о связи фармакологической активности со структурой вещества. В этом случае необходимы данные о пользе вещества или элемента.
Далее к избранному основному скелету вещества добавляют различные радикалы, которые будут способствовать растворению вещества в липидах и воде. Синтезируемую структуру целесообразно сделать растворимой одновременно и в воде, и в жирах с той целью, чтобы она могла всосаться в кровь, перейти из нее через гематотканевые барьеры в ткани и клетки и затем вступить в связь с клеточными мембранами или проникнуть через них внутрь клетки и соединиться с молекулами ядра и цитозоля [6].
Целенаправленный синтез лекарственных веществ становится удачным, когда удается найти такую структуру, которая по размеру, форме, пространственному положению, электронно-протонным свойствам и ряду других физико-химических показателей будет соответствовать живой структуре, подлежащей регулированию.
Целенаправленный синтез веществ преследует не только практическую цель - получение новых лекарственных веществ с нужными фармакологическими и биологическими свойствами, но и является одним из методов познания общих и частных закономерностей жизненных процессов. Для построения теоретических обобщений необходимо дальнейшее изучение всех физико-химических характеристик молекулы и выяснение решающих изменений в ее структуре, обусловливающих переход одного вида активности в другой.
Составление комбинированных препаратов является одним из наиболее эффективных путей поиска новых лекарственных средств. Принципы, на основе которых восставляются многокомпонентные лекарственные препараты могут быть различными и изменяются вместе с методологией фармакологии [7]. Разработаны основные принципы и правила составления комбинированных средств.
Чаще всего в комбинированные средства включаются лекарственные вещества, которые оказывают действие на этиологию заболевания и основные звенья патогенеза болезни. В комбинированное средство обычно включаются лекарственные вещества в малых или средних дозах, если между ними существуют явления взаимного усиления действия (потенцирование или суммирование).
Комбинированные средства, составленные с учетом указанных рациональных принципов, отличаются тем, что они вызывают значительный лечебный эффект при отсутствии или минимуме отрицательных явлений. Последнее их свойство обусловлено введением малых доз отдельных ингредиентов. Существенное преимущество малых доз состоит и в том что они не нарушают естественных защитных или компенсаторных механизмов организма.
Комбинированные препараты составляются также и по принципу включения в них таких дополнительных ингредиентов, которые устраняют отрицательное действие основного вещества.
Комбинированные препараты составляются с включением различных корригирующих средств, устраняющих нежелательные свойства основных лекарственных веществ (запах, вкус, раздражение) или регулирующих скорость освобождения лекарственного вещества из лекарственной формы или скорость всасывания его в кровь.
Рациональное составление комбинированных средств позволяет целенаправленно увеличить фармакотерапевтический эффект и устранить или уменьшить возможные отрицательные стороны действия лекарственных средств на организм.
При комбинировании лекарственных средств отдельные компоненты должны быть совместимы между собой в физико-химическом, фармакодинамическом и фармакокинетическом отношениях [8].
1.3 Роль компьютера при создании новых лекарственных средств
Ежегодно химики синтезируют, выделяют и характеризуют от 100 до 200 тысяч новых веществ. Многие из этих веществ проходят первичные испытания на выявление той или иной биологической активности. Этот этап поиска лекарственного вещества называют скринингом. Скрининг проводят в биологических лабораториях на живых клетках, микроорганизмах или кусочках живых тканей, на здоровых или специально задержанных животных: на мышах, крысах, морских свинках, собаках.
При этом из сотен веществ отбираются несколько наиболее активных препаратов, которые затем передаются на углубление испытания. Если высокая активность вещества подтверждается, то его всесторонне изучают для определения токсичности и побочных эффектов, при отсутствии или незначительности которых проводят кинетические испытания на людях.
Считается необходимым, чтобы все новые синтезируемые вещества были подвергнуты первичным испытаниям. Очевидно, что возможность испытать все новые соединения на все новые соединения на все нужные виды активности пока остается малореальной. В настоящее время существует возможность определения потенциала их биоактивности путем компьютерного анализа [1]. Достаточно лишь ввести в компьютер сведения о строении вещества. По окончании компьютерного анализа оператор получает рекомендации о целесообразности или нецелесообразности испытаний данного вещества на тот или иной вид активности. Скрининг экономит время, материалы и силы при аналоговом поиске лекарственных веществ.
В настоящее время также пользуются методом химического модифицирования структуры известных синтетических и природных лекарственных веществ. Этот метод является интуитивным, умозрительным. С его помощью исходя из аналогии двух структур биоактивность известного вещества как бы переносят на новое соединение.
Метод молекулярного моделирования в сочетании с рентгеноструктурным анализом позволяет установить стехереохимические особенности молекулы лекарственного вещества и биорецептора, конфигурацию их хиральных центров, измерить расстояние между отдельными атомами, группами атомов или между зарядами в случае цвиттер-ионных структур лекарства и биорецепторного участка его захвата. Получаемые таким образом данные позволяют более целенаправленно проводить синтезы биоактивных молекул с заданными на молекулярном уровне параметрами. Этот метод был успешно использован в синтезе высокоэффективных анальгетиков – аналогов морфина, а также для получения ряда лекарственных веществ, действующих на центральную нервную систему подобно природному нейромедиатору γ – аминомасляной кислоты. Широкое развитее получил метод комбинаторной химии.
Метод комбинаторной химии возник и стал быстро развиваться в 1990-х годах, как часть общей стратегии открытия новых лекарственных веществ.
Стратегия комбинаторной химии основана на недавней разработке нескольких революционных химических и биологических методов параллельного синтеза и испытания большого числа соединений. Была создана техника, позволяющая синтезировать в растворе или на твердых подложках от сотен до нескольких тысяч новых соединений в день и быстро их тестировать в виде смесей или после выделения индивидуальных веществ. В совокупности с автоматизацией синтез целых семейств вещества требует значительно меньше затрат реагентов при огромном росте производительности [9].
1.4 Молекулярное моделирование с помощью программы HyperChem
Молекулярное моделирование – сложная сеть различных наук, находящее применение в нанотехнологии, в молекулярной биологии, квантовой химии и биотехнологии.
Молекулярное моделирование молодая, востребованная и бурно развивающаяся наука.
На сегодняшний день методы квантовой химии и молекулярной динамики получили широкое распространение в численном моделировании электронной и атомной структур сложных систем молекулярных, кристаллических и переходных размеров. Это связано с технологическим развитием соответствующего математического обеспечения. Сейчас в мире функционирует достаточно много современных вычислительных комплексов, реализующих методы квантовой химии и молекулярной динамики. Использование многих из этих методов обеспечивается программой Hyper Chem для молекулярного моделирования.
HyperChem - комплексный программный продукт, предназначенный для задач молекулярного моделирования. Он включает в себя программы, реализующие методы молекулярной механики, квантовой химии и молекулярной динамики. Силовые поля, которые могут использоваться в HyperChem - это ММ+ (на базе ММ2), Amber, OPLS и BIO+ (на базе CHARMM). Реализованы полуэмпирические методы: расширенный метод Хюккеля, CNDO, INDO, MINDO/3, MNDO, AM1, PM3, ZINDO/1, ZINDO/S, а также возможности проведения неэмпирических расчетов и по теории возмущений Меллера-Плессета второго порядка.
HyperChem обладает развитыми средствами визуализации, которые могут использоваться как при подготовке входной информации (структуры молекулы), так и при анализе результатов, например, рассчитанных характеристик ИК- и УФ- спектров.
Расчётные методы оказывают неоценимую помощь в создании лекарственных средств. Молекулярное моделирование входит во все области знаний и находит себе применение, порой играя одну из главных ролей. Некоторые области химии немыслимы без молекулярного моделирования. В развитых странах моделирование является современным методом изучения микроструктур.
В настоящее время для изучения реакционной способности молекул используются приближения CNDO/2, MNDO, AM1, PM3.
Метод CNDO основан на приближении нулевого дифференциального перекрывания и поэтому является одним из простейших полуэмпирических методов. Из этого факта следуют ограничения применимости метода, который из-за обедненной расчетной схемы недостаточно корректно воспроизводит многие эффекты. С появлением более совершенных версий полуэмпирических методов МО приближение CNDO все реже применяется на практике. Так, в версии 7 программного продукта МОРАС данный метод не представлен. Тем не менее, во многих случаях для быстрой оценки электронных параметров полезно использовать схему CNDO, так как вследствие резкого уменьшения количества рассчитываемых интегралов с помощью этого метода можно исследовать более сложные объекты. В целом CNDO/2 дает надежные результаты при расчете электронных распределений и свойств, зависящих от них.
Основным калибровочным параметром в CNDO является резонансный интеграл. Он подбирается так, чтобы относительный порядок энергетических уровней занятых МО и коэффициенты разложения МО в ЛKAO наилучшим образом совпали с расчетами ab initio соединений обучающей выборки.
Общим достоинством всех перечисленных версий является прежде всего сравнительно малое время расчетов и меньшие размеры занимаемой оперативной памяти по сравнению с более точными приближениями. Это дает возможность как для быстрой оценки исследуемых объектов, так и для изучения более сложных молекул, требующих длительного времени расчета и больших объемов оперативной памяти. В целом приближение CNDO хорошо описывает электростатические эффекты и полярность связи. CNDO/2 может применяться для расчета дипольных моментов и зарядов по схеме Малликена и оценки равновесной геометрии.
Недостатки приближения CNDO являются следствием усечения расчетной схемы, которая не учитывает взаимодействия между перекрываниями орбитальных зарядов. В результате многие эффекты не воспроизводятся.
Метод MNDO был разработан на основе более строгого и сложного приближения NDDO. Это позволило существенно улучшить результаты расчетов при решении многих задач. Длительное время метод рассматривался в качестве основного полуэмпирического метода квантовой химии. Его возможности позволили с достаточной степенью надежности рассчитывать физико-химические свойства, электронные структуры и реакционную способность множества молекулярных систем.
Преимущество заключается в быстродействии (по сравнению с неэмпирическими методами) программ, в которых реализована схема MNDO. Это позволяет применять ее для исследования все более сложных объектов. Недостатки связаны с тем, что точность метода не может превышать точность тех экспериментальных данных, по которым проводилась параметризация.
В схеме MNDO используются 3 вида параметров.
Во-первых, варьируемые параметры, значения которых определяют с помощью оптимизационной процедуры.
Во-вторых, одноцентровые двухэлектронные интегралы, оценивающиеся из спектроскопических данных.
В-третьих, ряд зависимых параметров, необходимых для расчета двухцентровых двухэлектронных интегралов, оценивающиеся с помощью эмпирических схем.
В настоящее время область применения метода MNDO достаточно изучена. Зная особенности расчетной схемы MNDO, его преимущества и недостатки можно с успехом применять метод для решения многих задач.
Хотя в целом метод MNDO имеет существенные преимущества перед СNDO, в некоторых случаях метод дает серьезные сбои. Это в первую очередь касается расчетов молекул с водородными связями, барьеров внутреннего вращения в p-сопряженных системах и расчетов четырехчленных циклов. Поэтому в рамках метода MNDO были разработаны модифицированные варианты.
Для расчета характеристик систем с водородными связями были разработаны методы MNDO/Н и MNDO/М, которые лучше воспроизводят экспериментальные значения энергии водородных связей в комплексах.
Удовлетворительное описание водородных связей позволило широко использовать модифицированные варианты для исследования биологических объектов.
Таким образом, полуэмпирические квантовохимические методы можно использовать с большим практическим выходом для изучения реакционной способности различных химических соединений.
| Достоинства метода MNDO |
Недостатки метода MNDO |
| 1.Быстродействующий метод, позволяющий изучать строение и свойства сложных молекул. 2.Учитывается ориентация р-орбиталей и правильно описывается отталкивание неподеленных электронных пар. 3.Значительное расширение круга доступных для расчета соединений. 4.Более корректное описание последовательности верхних молекулярных уровней. 5.Может использоваться для интерпретации фотоэлектронных спектров. 6.Преимущества проявляются в большей степени в расчетах более полярных молекул. 7.Удовлетворительно описывается строение радикалов, дает разумные результаты для катионов органических соединений. 8.Более точно рассчитываются валентные углы. 9.Более надежный расчет частот колебаний, протонных эффектов и электронного сродства. 10.При изучении химических реакций правильно описывается ППЭ и ПС. |
1.Точность метода не может превышать точности экспериментальных данных. 2.Электронная корреляция учитывается дважды. Правильнее было бы внесение корреляционных поправок. 3.Недооцениваются эффекты взаимодействия через пространство. 4.Ограничения возможности расчета соединений третьего и более высоких периодов (в частности с SO и SO2-группами) вследствии пренебрежения d-орбиталями. 5.Некорректное описание водородных связей. 6.Неверен расчет барьеров внутреннего вращения в сопряженных молекулах. 7.Недооценивается энергия трехцентровых связей. 8.Неудовлетворительная оценка спиновых плотностей и констант сверхтонкого расщепления электронного парамагнитного резонанса. 9.Завышается стабильность радикалов. 10.Потенциалы ионизации атомов III периода с сильно выраженным S-характером завышены вследствие применения приближения замороженного остова. 11.Плохо воспроизводится спиновая плотность в радикалах. 12.Энтальпия одноатомных ионов может значительно отличаться от экспериментальных данных. 13.Ограниченная воспроизводимость свойств неорганических молекул. Ошибка расчета составляет для энтальпии - 10 ккал/моль, потенциала ионизации - 1 эВ, длин связей - 0,07A |
1.5 Определение спектра биологической активности с помощью программы PASS C & T ( Prediction of Activity Spectra for Substances : Complex & Training )
Современная версия компьютерной системы предсказания спектра биологической активности PASS C&T (P rediction of A ctivity S pectra for S ubstances: C omplex & T raining) реализована в 1998 году. Она включает в себя обучающую выборку, содержащую более 45000 биологически активных веществ с известной биологической активностью, и охватывает более 400 фармакологических эффектов, механизмов действия, а также мутагенность, канцерогенность, тератогенность и эмбриотоксичность.
Работа PASS основана на анализе зависимостей «структура-активность» для веществ из обучающей выборки, содержащей более 45000 разнообразных биологически активных веществ (субстанции известных лекарственных препаратов и фармакологически активные соединения). Обучающая выборка постоянно пополняется новой информацией о биологически активных веществах, отбираемой как из публикаций в научно-технической литературе, так и из многочисленных баз данных. Химическая структура представлена в PASS в виде оригинальных MNA дескрипторов (Mulilevel Neighbourhoods of Atoms). MNA дескрипторы имеют универсальный характер и с достаточно хорошей точностью описывают разнообразные зависимости «структура-свойство». Используемый в PASS математический алгоритм был отобран путем целенаправленного анализа и сравнения эффективности для решения подобных задач большого числа различных методов. Показано, что данный алгоритм обеспечивает получение устойчивых в статистическом смысле зависимостей “структура-активность” и, соответственно, результатов прогноза. Это очень важно, поскольку включенные в обучающую выборку данные всегда обладают определенной неполнотой как в отношении охвата всех химических классов веществ, имеющих конкретный вид активности, так и в отношении изученности каждого отдельного вещества на все возможные виды активности. Средняя точность прогноза при скользящем контроле составляет свыше 85%. Скользящий контроль проводится следующим образом: из обучающей выборки поочередно удаляется одно вещество и для него делается прогноз на основе анализа оставшейся части обучающей выборки, результат сравнивается с известными экспериментальными данными. Процедура повторяется итеракивно для каждого из веществ и рассчитывается средняя точность прогноза. Точность прогноза в 85% достаточна для практического применения системы PASS с целью прогноза спектра биологической активности новых веществ, поскольку ожидаемая вероятность случайного угадывания одного из 780 видов активности составляет около 0.1% [10].
Результаты прогноза выдаются пользователю в виде списка названий вероятных видов активности с расчетными оценками вероятностей наличия (Pa) и отсутствия каждого вида активности (Pi), которые имеют значения от 0 до 1. Эти вероятности рассчитываются независимо по подвыборкам активных и неактивных соединений, и поэтому их сумма не равна единице. Pa и Pi интерпретируются как оценки меры принадлежности вещества к классам активных и неактивных соединений соответственно, либо как оценки ошибок первого и второго рода. Чем больше для конкретной активности величина Pa и чем меньше величина Pi, тем больше шанс обнаружить данную активность в эксперименте. Если при анализе прогнозируемого списка активностей для исследования выбираются те виды активности, для которых Pa>90%, то мы рискуем пропустить около 90% действительно активных соединений, но вероятность ложноположительных прогнозов при этом ничтожно мала; для Pa>80% - пропустим уже только 80% активных соединений, но и вероятность ложноположительных прогнозов будет выше, наконец, для Pa>Pi вероятности ошибок первого и второго рода равны [1, 9, 10].
На практике, однако, при отборе для исследования наиболее перспективных веществ руководствуются и другими критериями, например, критерием новизны. При этом исходят из того, что чем ближе значение Pa к единице, тем более вероятно, что вещество является близким аналогом известного препарата. Поэтому, если целью исследователя является выявление соединений с достаточно высоким уровнем новизны (New Chemical Entity, NCE), то надо выбирать вещества, для которых величина прогнозируемой вероятности Pa для требуемого вида активности несколько ниже, например, 0,5<PA< NCE.
Базируясь на данных компьютерного прогноза, исследователь может:
· определить, какие тесты наиболее адекватны для изучения биологической активности конкретного химического соединения.
· обнаружить новые эффекты и механизмы действия для ранее изученных веществ;
· отобрать наиболее вероятные базовые структуры новых лекарств с требуемым биологическим действием среди доступных для скрининга химических соединений.
Система PASS позволяет получить прогноз спектра биологической активности 1000 веществ на обычном персональном компьютере менее чем за одну минуту. Поскольку прогноз выполняется по структурной формуле вещества, он может быть выполнен уже на стадии планирования синтеза [11].
Применимость PASS для решения практических задач продемонстрирована в многочисленных экспериментах. Прогнозируемые виды активности подтверждены для веществ различных химических классов, проявляющих разнообразные эффекты: противобактериальный, антиаритмический, противоопухолевый, гепатопротекторный, антиамнестический, противоспалительный, антиоксидантный и др.
С применением PASS при поддержке гранта CRDF (RC1-2064) был выполнен прогноз спектра биологической активности для 250000 химических соединений.
Кроме того, если, наряду с основным действием, известен перечень нежелательных побочных эффектов, то при отборе перспективных для исследований соединений можно руководствоваться комбинированным критерием:
· наличие в прогнозируемом спектре требуемых эффектов/механизмов;
· отсутствие нежелательных эффектов/механизмов.
Естественно, что при рассмотрении всего списка, включающего свыше 400 прогнозируемых PASS C&T видов активности, можно составить большое количество комбинаций из требуемых и нежелательных эффектов.
Для их анализа сотрудник Лаборатории структурно-функционального конструирования лекарств НИИ Биомедхимии РАМН А. А. Лагунин разработал специальную компьютерную систему интерпретации спектров биологической активности веществ IBIAC , основанную на знаниях об известных взаимосвязях между фармакологическими эффектами и механизмом действия биологически активных веществ (более 2000 терминов, описывающих биологическую активность).
С использованием системы IBIAC генерация перечня эффектов, соответствующих определенному механизму действия и, наоборот, списка вероятных механизмов, ответственных за проявление определенного эффекта, осуществляется автоматически [12].
Поскольку прогноз спектра биологической активности осуществляется на основе структурной формулы химического соединения, он может быть выполнен уже на этапе планирования синтеза.
В итоге будут синтезированы лишь некоторые из теоретически возможных производных, в наибольшей степени удовлетворяющие критериям задачи.
Необходимо отметить, что прогноз спектра биологической активности возможен для низкомолекулярных органических (drug-like) соединений, структура которых не отличается принципиально от веществ обучающей выборки. Не имеет смысла прогноз для синтетических и биополимеров, для неорганических веществ и т.п [13, 14, 15, 16].
1.6 Местноанестезирующие средства
Несмотря на достижения современной анестезии, продолжаются поиски менее опасных средств для наркоза, разработка различных вариантов многокомпонентного избирательного наркоза, позволяющего значительно снизить дозы используемых средств, уменьшить их токсичность и побочные отрицательные влияния. Известно, что местноанестезирующие средства понижают чувствительность окончаний афферентных нервных волокон, и угнетают проведения возбуждения по нервным волокнам. Вызывают местную потерю чувствительности. И в первую очередь они устраняют чувство боли, в связи с чем их используют главным образом для местного обезболивания (анестезии) [17].
К анестезирующим средствам предъявляют определенные требования. Прежде всего они должны иметь высокую избирательность действия, не оказывая отрицательного влияния ни на нервные элементы, ни на окружающие ткани. Короткий латентный период, высокая эффективность при разных видах местной анестезии, определенная продолжительность действия. Желательно, чтобы они сужали кровеносные сосуды. Это существенный момент, так как сужение сосудов усиливает анестезию, понижает кровотечение из тканей, а так же уменьшает возможность токсических эффектов задерживая всасывание анестетика. Так же к числу важных характеристик относятся низкая токсичность и минимальные побочные эффекты.
В настоящее время в медицинской практике используется множество местноанестезирующих веществ с различной степенью активности и разной продолжительностью действия.
По применения в клинической практике местные анестетики подразделяются на:
1) средства, применяемые только для поверхностной анестезии: кокаин, дикаин (тетракаин), бензокаин (анестезин), бупивакаин (пиромекаин);
2) средства, применяемые для инфильтрационной и проводниковой анестезии: новокаин (прокаин), тримекаин, бутивокаин, изокаин, ультракаин.
3) средства, применяемые для всех видов анестезии: лидокаин (кскаин).
По химическому строению местноанестезирующие вещества можно разделить на две группы:
1) сложные эфиры: кокаин, дикаин, бензокаин, прокаин;
2) замещенные амиды кислот: лидокаин, тримекаин, бупивакаин, булекаин.
Дикаин применяется для поверхностной анестезии. Под влиянием местных анестетиков в окончании нерва и в самом нерве прекращается электрохимический процесс, осуществляющий передвижение ионов через мембрану и распространение нервных импульсов. Область окончания чувствительных нервов находится под регулирующим влиянием системы медиаторных рецепторов, синергентное взаимодействие, которых обеспечивает более эффективное развитее торможения.
Так же известно, что в присутствии противогистаминных средств (димедрол), м – холиноблокаторов (атропин) и адренанила усиливается местноанестезирующий эффект [18].
Дикаин представляет собой ![]() - диметиламиноэтиловй эфир п – бутиламинобензойной кислоты гидрохлорид).
- диметиламиноэтиловй эфир п – бутиламинобензойной кислоты гидрохлорид).

Работая над компьютерным дизайном местноанестезирующего препарата дикаин, был проведен информационный анализ данного препарата по сравнению с другими средствами, такими как бензокаин (анестезин), бумекаин. В ходе анализа было установлено, что дикаин является эффективным местноанестезирующим средством, значительно превосходящим по активности кокаин (примерно в 10 раз), он также превосходит кокаин по токсичности (в 2-5 раз).
Применяют дикаин в глазной практике в виде 0,1 % раствора при измерении внутриглазного давления, в виде 0,25-1% или 2% раствора при удалении инородных тел. Дикаин в отличие от кокаина не влияет на внутриглазное давление, не расширяет зрачки. При необходимости длительной анестезии используют глазные пленки с дикаином (0,75 см) изготовленные на основе биорастворимого полимера.
Анестезиофорной группой является диалкиламиноацетанилид. Расстояние между ![]() и
и ![]() атомами определяет двухточечный контакт молекулы дикаина с рецептором через диполь-дипольное и ионное взаимодействие. В структуру молекулы дикаина входит фрагмент биогенного вещества коламина, производные которого оказывают противогистаминный эффект. Дикаин активнее анестезина и новокаина, но токсичнее их в несколько раз. В проведении дальнейшего анализа были предложены варианты новых структур для компьютерного анализа.
атомами определяет двухточечный контакт молекулы дикаина с рецептором через диполь-дипольное и ионное взаимодействие. В структуру молекулы дикаина входит фрагмент биогенного вещества коламина, производные которого оказывают противогистаминный эффект. Дикаин активнее анестезина и новокаина, но токсичнее их в несколько раз. В проведении дальнейшего анализа были предложены варианты новых структур для компьютерного анализа.
2. Методическая часть
2.1 Характеристика объекта исследования
Температура плавления 147-150![]() . Соединение представляет белый кристаллический порошок без запаха. Легко растворим в воде и спирте. Дикаин сильное местноанестезирующее средство, обладающее высокой токсичностью. Применяют в глазной и оториноларингологической практике при некоторых оперативных вмешательствах, а также для анестезии.
. Соединение представляет белый кристаллический порошок без запаха. Легко растворим в воде и спирте. Дикаин сильное местноанестезирующее средство, обладающее высокой токсичностью. Применяют в глазной и оториноларингологической практике при некоторых оперативных вмешательствах, а также для анестезии.
2.1. 1 Характеристика приборов
Методы исследования проводили с помощью программы Hyper Chem.
2.1. 2 Методика выполнения
Построить молекулы при помощи программы HyperChem.
Последовательность действия:
1. Запуск программы Hyper Chem.
2. Создание молекулы:
- мышь устанавливают на пункт меню “Build”, щелчком по левой кнопке разворачивают меню и выбирают “Default atoms”;
- устанавливают указатель курсора в режиме построения молекулярных моделей; для этого в верхней строчке выбирают необходимый вид курсора и щелкают левой кнопкой мышки;
- в развёрнутой на экране периодической таблице выбирают интересующий атом;
- связь между атомами обозначают, нажимая левую кнопку мыши в положение одного из атомов, и, удерживая ее, передвигают курсор к другому атому; затем кнопку отпускают, типы связей задают. Полуторные связи ароматического кольца обозначаются пунктирной линией. Их можно нарисовать при помощи двойного L-щелчка вблизи одной из внутренних сторон кольца;
- построенную модель автоматически дополняют атомами водорода; для этого мышь устанавливают на пункт меню “Build”, щелчком по левой кнопке разворачивают меню и выбирают “Add Hydrogens”.
- для устранения неточностей выполненного рисунка мышь устанавливают на пункт меню “Build”, щелчком по левой кнопке разворачивают меню и выбирают “Model build”; данная команда корректирует межатомные расстояния и углы.
С помощью программы Нурег Chem:
- курсор мыши устанавливают на пункт меню "Setup", выбирают метод молекулярной механики), устанавливают "ММ";
- запускают процесс оптимизации геометрии путем выбора пункта меню "Compute", далее выбирают "Geometry Optimize";
Определение геометрических характеристик (длин связей и валентных углов):
- выбирают курсор в виде двух концентрических окружностей, ставят этот курсор на один из интересующих атомов, нажимают левую кнопку мыши и, не отпуская ее, подводят курсор к следующему атому (для измерения длины связи) или к атому, находящемуся через один от исходного (для измерения величины валентного угла); затем кнопку отпускают.
В нижней строке экрана появится значение длины связи (Е) или валентного угла (град.).
- в пункте меню "File, выбирают "Start Log" (создание файла отчета); файлу дают название и устанавливают "Quantum print level" = 9;
- в пункте меню "Setup", выбирают "Semiempirical methods"; в раскрывшемся окошечке устанавливают "MNDO";
- устанавливают соответствующий заряд и мультиплетность в соответствующих полях;
- запускают процесс расчета с оптимизацией геометрии путем выбора пункта меню "Compute", выбирают "Geometry Optimize";
- расчет заканчивается, когда в нижней строке окна появляется надпись Conv=YES"- закрывают файл отчета (.log file) путем выбора пункта меню "File", выбирают "Stop Log".
Графическое изображение ВЗМО и НВМО.
- для получения графического изображения молекулярной орбитали выбирают пункт меню "Compute", выбирают "Orbitals";
- выбирают номер нужной молекулярной орбитали, и устанавливают "3D";
- полученную картинку можно скопировать, используя пункт меню "Edit" и далее "Copy image".
- выбирают пункт меню "Compute", затем "Plot molecular properties";
- выбирают "electrostatic potencial" и устанавливают "3D". Положительный знак электростатического потенциала отображается зеленым цветом. В области неподеленных пар на атомах азота, кислорода и др. электростатический потенциал отрицательный, что отображается красным цветом.
- выделить в нарисованной молекуле связь;
- в меню Comput и выбрать пункт Potencial;
- задать начальную(Initial Bond Angle), конечную(Final Bond Angl) длины связи и шаг (Step) с которым будут автоматически производиться расчеты.
-после расчетов машина построит на экране график изменения потенциальной энергии выбранной связи от величины растяжения.
2.2 Определение биологической активности молекул с помощью программы PASS
2.2. 1 Методика выполнения
Построить молекулы при помощи программы Isis Draw.
Последовательность действия:
1. Запуск программы Isis Draw.
2. Создание молекулы:
- мышь устанавливают на клавишу “Single Bound” для того, что бы изобразить фрагмент связи;
- устанавливают указатель курсора на клавишу “Atom” для того, что бы выбирать интересующий атом;
- построенную модель сохраняют, устанавливая курсор на пункт меню “File”, затем “Export”, устанавливают на “Molfile” и сохраняют в директорию Isis Draw.
Последовательность действия:
1. Запуск программы PASS.
2. Исследование биологической активности:
- мышь устанавливают на пункт меню “File”, затем “Open Base” выбираем “passdemo.SAR” и нажимаем “Открыть”;
- устанавливают указатель курсора на клавишу “Predict and Save Prediction as Text files”, тип файлов выбираем “mol”, нажимаем “Открыть”, затем сохраняем как “txt”, открываем сохраненный файл и изучаем биологическую активность.
3. Результаты эксперимента и их обсуждение
3.1 Разработка новых молекулярных структур на основе дикаина
Несмотря на достижения современной анестезии, продолжаются поиски менее опасных средств для наркоза, разработка различных вариантов многокомпонентного избирательного наркоза, позволяющего значительно снизить дозы используемых средств, уменьшить их токсичность и побочные отрицательные влияния [19]. В последнее время методы компьютерного моделирования все более входят в практику технологии создания новых синтетических лекарственных веществ [9]. Этот подход позволяет установить стехиометрические особенности молекулы лекарственного соединения, измерить расстояние между отдельными атомами, определить потенциал биоактивности, комплементарность взаимодействия с биорецепторным участком. Получаемые таким образом данные позволяют более целенаправленно проводить синтезы биоактивных молекул с заданными на молекулярном уровне параметрами, что значительно экономит время, материалы и силы при аналоговом поиске лекарственных веществ.
Дикаин применяется для поверхностной анестезии. Под влиянием местных анестетиков в окончании нерва и в самом нерве прекращается электрохимический процесс, осуществляющий передвижение ионов через мембрану и распространение нервных импульсов. Область окончания чувствительных нервов находится под регулирующим влиянием системы медиаторных рецепторов, синергентное взаимодействие, которых обеспечивает более эффективное развитие торможения. Известно, что в присутствии противогистаминных средств (димедрол), м- холиноблокаторов (атропин) и адреналина усиливается местноанестезирующий эффект [20].
Дикаин относится к классу сложных эфиров п -аминобензойной кислоты (β-диметиламиноэтиловый эфир п -бутиламинобеизойной кислоты гидрохлорид) [8].

Анестезиофорной группой является диалкиламиноацетанилид. Расстояние между ![]() и
и ![]() атомами определяет двухточечный контакт молекулы дикаина с рецептором через диполь-дипольное и ионное взаимодействие. В структуру молекулы дикаина входит фрагмент биогенного вещества коламина (2-аминоэтанола), производные которого оказывают противогистаминный эффект. Дикаин активнее своих аналогов (анестезина и новокаина), но и токсичнее их в несколько раз. Его используют главным образом в глазной и оториноларингологической практике.
атомами определяет двухточечный контакт молекулы дикаина с рецептором через диполь-дипольное и ионное взаимодействие. В структуру молекулы дикаина входит фрагмент биогенного вещества коламина (2-аминоэтанола), производные которого оказывают противогистаминный эффект. Дикаин активнее своих аналогов (анестезина и новокаина), но и токсичнее их в несколько раз. Его используют главным образом в глазной и оториноларингологической практике.
Нами предложены варианты новых структур для компьютерного дизайна молекулы дикаина с целью снижения его токсичности с сохранением или даже усилением анестезирующих свойств.
Введение в бензольное кольцо «облагораживающей» карбоксильной группы и замена диметиламиногруппы на более фармакоактивную диэтиланиногруппу позволит снизить токсичность соединения, облегчить гидролиз сложноэфирной связи с высвобождением антигистаминного фрагмента - диэтиламиноэтанола.
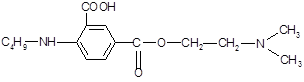
Алифатический радикал н -бутил в структуре дикаина усиливает фармакологический эффект. При замене его на адреналиновый фрагмент ожидается получить более яркое анестезирующее действие.

К настоящему времени известно, что биологические системы не делают различия между плоскими кольцами, поэтому при замене ароматической основы н -аминобензойной кислоты на никотиновую (или изоникотиновую) кислоту изменяется полярность молекулы, облегчается задача введения различных заместителей в ароматическое кольцо. К тому же, аминопроизводные никотиновой кислоты (кордиамин) являются стимуляторами центральной нервной системы.

Один из наиболее эффективных анестетиков, промедол, содержит в структуре вместо ароматического пиридинового кольца пиперидиновое, что является предпосылкой для модификации молекулы дикаина.

3.2 Результаты исследований с помощью программы HyperChem
Таблица 1
| Длина связи или валентный угол |
Данные ММ расчёта |
Данные MNDO расчёта |
Справочные величины |
| С1-С2 С2-С3 С3-С4 С4-N5 N5-C6 C6-C7 C7-C9 C9-C11 C10-C11 C8-C10 C6-C8 C11-C12 C12-O13 C12-O14 O14-C15 C15-C16 C16-N17 N17-C19 N17-C18 C1-C2-C3 С2-С3-С4 C3-C4-N5 N5-C6-C8 N5-C6-C7 C6-C7-C9 C7-C9-C11 C9-C11-C10 C11-C10-C8 C10-C8-C6 C8-C6-C7 C9-C11-C12 C10-C11-C12 C12-O14-C15 O14-C15-C16 C15-C16-N17 C16-N17-C18 C16-N17-C19 C18-N17-C19 |
1,53442 1,53705 1,53626 1,44712 1,35045 1,34373 1,34335 1,34525 1,3446 1,34261 1,34354 1,36294 1,21202 1,35065 1,40995 1.53635 1,45672 1,45174 1,45291 111,792 113,518 109,67 118,219 123,605 120,397 120,124 119,658 119,913 120,666 119,242 119,948 122,115 118,556 107,759 113,916 111,47 113,282 111,148 |
1,53158 1,54128 1,54862 1,46777 1,41076 1,41935 1,40351 1,41482 1,41571 1,40009 1,42528 1,49637 1,22988 1,36302 1,41167 1,56222 1,47112 1,46359 1,46386 114,547 113,518 111,073 118,219 123,605 120,589 121,461 117,898 121,164 120,879 117,974 119,948 122,115 124,819 107,759 109,436 116,806 117,015 116,056 |
1,534 1,536 1,539 1,456 1,411 1,393 1,355 1,381 1,392 1,356 1,352 1,383 1,218 1,386 1,382 1,559 1,493 1,432 1,451 113,125 113,529 110,053 118,953 123,983 120,159 120,956 118,241 120,971 120,264 118,325 119,948 122,233 121,179 107,563 110,563 115,963 115,912 116,023 |
Сравнивая полученные результаты, оба метода имеют небольшие отклонения по сравнению со стандартными данными.
Расположение молекулы немного изменяется в зависимости от применяемого метода.
Положительный знак электростатического потенциала отображается зелёным цветом. В области неподелённых пар на атомах азота, кислорода электрический потенциал отрицательный, что отображается красным цветом.
Изменение потенциальной энергии связи C16-N17.
Исследуемая связь между атомами C16-N17. Задаём начальные величины начальная длина связи 0,971; конечная длина связи 2,971; шаг 0,1. Проведя расчёты, изобразим график изменения потенциальной энергии связи C16-N17 от величины растяжения.
Изменение потенциальной энергии углового напряжения C3-C4-C5.
Исследуем угол между атомами C3-C4-C5. Задаём начальные величины начальный угол 80; конечный 140; шаг 10. Проведя расчёты, изобразим график изменения энергии углового напряжения при изменении величины угла.
Таблица 2
| Длина связи или валентный угол |
Данные ММ расчёта |
Данные MNDO расчёта |
Справочные величины |
| C1-C2 C2-C3 C3-C4 C4-N5 N5-C6 C6-C7 C7-C9 C9-C10 C10-C11 C11-C8 C6-C8 C7-C12 C12-O13 C12-O14 C10-C15 C15-O16 C15-O17 O17-C18 C18-C19 C19-N20 N20-C23 C23-C24 N20-C21 C21-C22 C1-C2-C3 C2-C3-C4 C3-C4-N5 C4-N5-C6 N5-C6-C7 N5-C6-C8 C6-C7-C12 C7-C12-O13 C7-C12-O14 C6-C7-C9 C7-C9-C10 C9-C10-C11 C10-C11-C8 C6-C8-C11 C8-C6-C7 C11-C10-C15 C9-C10-C15 C10-C15-O16 C10-C15-O17 O16-C15-O17 C15-O17-C18 O17-C18-C19 C18-C19-N20 C19-N20-C21 C19-N20-C23 N20-C23-C24 N20-C21-C22 |
1,53446 1,53687 1,53769 1,46773 1,35455 1,34989 1,34842 1,3461 1,34288 1,34136 1,34515 1,36573 1,20944 1,33807 1,36377 1,21204 1,35143 1,41104 1,53791 1,46268 1,45983 1,53447 1,46076 1,53704 111,785 112,138 110,841 116,92 124,692 115,847 123,329 123,901 121,905 119,209 120,947 119,665 119,499 121,203 119,456 119,282 121,051 119,494 121,218 119,22 118,218 108,252 114,762 111,29 111,699 116,769 113,058 |
1,53154 1,54091 1,5495 1,47515 1,43364 1,423 1,41348 1,41233 1,41258 1,40248 1,41671 1,50204 1,22891 1,35818 1,50134 1,22774 1,35955 1,41312 1,5631 1,47233 1,47481 1,53447 1,47478 1,53801 111,785 113,622 110,841 119,144 118,391 122,522 121,784 126,174 144,416 119,516 121,22 118,829 120,542 120,848 118,99 121,044 120,117 125,793 113,418 120,418 125,334 107,378 110,057 116,349 116,243 112,593 112,593 |
1,532 1,539 1,544 1,483 1,423 1,312 1,342 1,401 1,362 1,341 1,395 1,502 1,211 1,348 1,551 1,221 1,355 1,413 1,531 1,452 1,453 1,534 1,460 1,538 111,785 112,953 110,841 117,526 117,318 117,286 122,463 123,901 144,411 119,291 121,231 118,973 119,513 121,203 119,361 120,964 121,051 120,169 119,432 120,111 119,213 107,314 112,953 113,561 114,246 111,548 112,987 |
Сравнивая полученные результаты, оба метода имеют небольшие отклонения.
Расположение молекулы немного изменяется в зависимости от применяемого метода.
Положительный знак электростатического потенциала отображается зелёным цветом. В области неподелённых пар на атомах азота, кислорода электрический потенциал отрицательный, что отображается красным цветом.
Изменение потенциальной энергии связи C6-С11.
Исследуемая связь между атомами C6-С11. Задаём начальные величины начальная длина связи 1,037; конечная длина связи 3,037; шаг 0,1. Проведя расчёты, изобразим график изменения потенциальной энергии связи C6-C11 от величины растяжения.
Изменение потенциальной энергии углового напряжения C6-C11-O14.
Исследуем угол между атомами C6-C11-O14. Задаём начальные величины начальный угол 90; конечный 150; шаг 10. Проведя расчёты, изобразим график изменения энергии углового напряжения при изменении величины угла.
Объектом является химическое соединение.
Таблица 3
| Длина связи или валентный угол |
Данные ММ расчёта |
Данные MNDO расчёта |
Справочные величины |
| O1-C2 C2-C3 C3-C4 C4-C5 C5-C7 C6-C7 C2-C6 C3-O8 C5-C9 C9-C10 C10-N11 N11-C12 N11-C13 C13-C14 C14-C15 C15-C16 C16-C18 C17-C18 C13-C17 C16-C19 C19-C20 C19-O21 O21-C22 C22-C23 C23-N24 N24-C25 N24-C26 O1-C2-C6 O2-C2-C3 C2-C3-C4 C3-C4-C5 C4-C5-C7 C5-C7-C6 C2-C6-C7 C3-C2-C6 O8-C3-C2 O8-C3-C4 C4-C5-C9 C7-C5-C9 C5-C9-C10 C9-C10-N11 C10-N11-C12 C13-N11-C12 C14-C13-N11 C17-C13-N11 C13-C14-C15 C14-C15-C19 C15-C16-C18 C16-C18-C17 C13-C17-C18 C17-C13-C14 C19-C16-C18 C19-C16-C15 O20-C19-C16 O21-C19-C16 O20-C19-O21 C19-O21-C22 O21-C22-C23 C22-C23-N24 C23-N24-C25 C23-N24-C26 C25-N24-C26 |
1,36051 1,34612 1,34441 1,3441 1,34399 1,34099 1,34297 1,36079 1,51602 1,54091 1,46417 1,45765 1,35257 1,3425 1,34235 1,34536 1,34524 1,34278 1,34206 1,36228 1,21202 1,35004 1,4099 1,53786 1,45675 1,45323 1,45163 120,802 120,483 119,222 122,579 117,432 120,669 121,368 118,714 120,184 120,594 121,181 121,385 115,402 114,762 123,704 116,621 120,198 119,72 120,071 119,877 120,004 119,927 120,013 120,082 119,383 120,612 119,581 120,826 119,469 119,174 110,679 113,688 111,433 113,311 111,143 |
1,35748 1,43712 1,4148 1,41582 1,41064 1,4048 1,41425 1,35924 1,51423 1,55179 1,47428 1,47004 1,43737 1,41646 1,40524 1,41186 1,41199 1,4048 1,41809 1,501 1,22707 1,36061 1,41166 1,5624 1,47048 1,46449 1,46417 123,264 118,062 119,488 121,583 118,032 121,572 120,637 118,674 117,63 122,882 121 120,916 117,262 113,931 116,04 114,642 123,731 117,39 120,376 120,696 118,949 120,676 120,373 118,878 120,928 120,123 126,438 112,643 120,919 125,616 107,249 109,754 116,591 116,835 115,996 |
1,359 1,381 1,368 1,388 1,347 1,358 1,413 1,362 1,514 1,551 1,469 1,462 1,395 1,561 1,395 1,354 1,456 1,400 1,346 1,447 1,210 1,369 1,469 1,537 1,475 1,462 1,495 122,645 119,472 119,265 121,583 117,463 120,873 121,145 118,714 118,139 121,246 121,012 121,213 114,786 113,746 122,612 115,621 120,198 118,365 120,376 120,519 119,789 120,581 120,373 120,566 120,491 120,573 122,469 117,721 120,651 123,125 108,452 110,956 117,854 114,651 113,786 |
Сравнивая полученные результаты, оба метода имеют небольшие отклонения.
Расположение молекулы немного изменяется в зависимости от применяемого метода.
Положительный знак электростатического потенциала отображается зелёным цветом. В области неподелённых пар на атомах азота, кислорода электрический потенциал отрицательный, что отображается красным цветом.
Изменение потенциальной энергии связи N11-С13.
Исследуемая связь между атомами N11-С13. Задаём начальные величины начальная длина связи 2,36; конечная длина связи 5,666; шаг 0,1. Проведя расчёты, изобразим график изменения потенциальной энергии связи N11-С13 от величины растяжения.
Изменение потенциальной энергии углового напряжения C19-О21-С22.
Исследуем угол между атомами C19-О21-С22. Задаём начальные величины начальный угол 70; конечный 130; шаг 10. Проведя расчёты, изобразим график изменения энергии углового напряжения при изменении величины угла.
Оптимизация геометрии и расчёт параметров молекулы методом молекулярной механики (ММ+ и MNDO метод).
Таблица 4
| Длина связи или валентный угол |
Данные ММ расчёта |
Данные MNDO расчёта |
Справочные величины |
| С1-С2 С2-С3 С3-С4 С4-N5 N5-C6 C6-C7 C7-C8 C41-C8 C9-C41 C5-C9 C7-C10 C10-O11 C10-O12 O12-C13 C13-C14 C14-N15 N15-C16 N15-C17 C1-C1-C3 C2-C3-C4 C3-C4-N5 C4-N5-C6 C4-N5-C9 N5-C6-C7 C6-C7-C8 C7-C8-C41 C8-C41-C9 C41-C9-N5 C9-N5-C6 C6-C7-C10 C8-C7-C10 C7-C10-C11 C7-C10-O12 C10-O12-O13 O12-C13-C14 C13-C14-N15 C14-N15-C16 C14-N15-C17 C16-N15-C17 |
1,53455 1,53762 1,5369 1,468 1,45685 1,5104 1,34294 1,34042 1,33896 1,35217 1,36168 1,21147 1,34976 1,41025 1,53651 1,45706 1,45297 1,4518 111,892 111,685 111,781 122,06 119,167 114,299 120,314 120,526 122,338 123,23 118,447 121,465 118,218 117,423 122,29 118,494 107,986 113,797 111,518 113,334 111,185 |
1,53153 1,54118 1,55042 1,47213 1,4767 1,51705 1,36463 1,44632 1,36782 1,39475 1,49687 1,22888 1,36285 1,41128 1,56218 1,47138 1,46379 1,46434 114,534 113,462 113,494 118,349 119,969 113,836 121,488 120,452 120,512 121,692 121,63 116,046 122,464 126,415 113,517 125,248 107,536 109,6 116,738 116,78 115,933 |
1,532 1,538 1,542 1,468 1,463 1,517 1,352 1,395 1,338 1,394 1,419 1,226 1,339 1,411 1,541 1,468 1,462 1,468 113,589 113,452 113,642 119,486 119,165 113,863 121,488 120,526 121,514 122,945 119,449 120,064 118,218 126,435 122,651 125,984 107,892 110,674 115,465 115,639 113,746 |
Сравнивая полученные результаты, оба метода имеют небольшие отклонения.
Расположение молекулы немного изменяется в зависимости от применяемого метода.
Положительный знак электростатического потенциала отображается зелёным цветом. В области неподелённых пар на атомах азота, кислорода электрический потенциал отрицательный, что отображается красным цветом.
Изменение потенциальной энергии связи С4–N5.
Исследуемая связь между атомами С4–N5. Задаём начальные величины начальная длина связи 0,972; конечная длина связи 2,972; шаг 0,1. Проведя расчёты, изобразим график изменения потенциальной энергии связи С4–N5 от величины растяжения.
Изменение потенциальной энергии углового напряжения C2-C3-С4.
Исследуем угол между атомами C2-C3-С4. Задаём начальные величины начальный угол 50; конечный 140; шаг 10. Проведя расчёты, изобразим график изменения энергии углового напряжения при изменении величины угла.
Оптимизация геометрии и расчёт параметров молекулы методом молекулярной механики (ММ+ и MNDO метод).
- Проведение оптимизации молекулы.
ММ+ метод.
Таблица 5
| Длина связи или валентный угол |
Данные ММ расчёта |
Данные MNDO расчёта |
Справочные величины |
| С1-С2 С2-С3 С3-С4 С4-N5 N5-C6 C6-C8 C8-C10 C9-C10 C7-C9 N5-C7 C10-C11 C11-O12 C11-O13 O13-C14 C14-C15 C15-N16 N16-C17 N16-C18 C1-C2-C3 C2-C3-C4 C3-C4-N5 C4-N5-C7 C4-N5-C6 N5-C7-C9 C7-C9-C10 C9-C10-C8 C6-C8-C10 C8-C6-N5 C6-N5-C7 C10-C11-O12 C9-C10-C11 C8-C10-C11 C10-C11-O13 O12-C11-O13 C11-O13-C14 C13-C14-C15 C14-C15-N16 C15-N16-C17 C15-N16-C18 |
1,53461 1,53838 1,53856 1,45536 1,45232 1,53586 1,53611 1,53558 1,536 1,4525 1,52029 1,20871 1,34376 1,40919 1,53584 1,45663 1,45283 1,45193 111,827 111,685 116,445 113,912 113,888 111,468 110,753 108,473 112,918 111,794 116,304 127,3 110,877 112,884 112,782 119,917 125,512 107,339 109,614 116,591 116,809 |
1,53147 1,54129 1,55152 1,47019 1,46727 1,54676 1,54834 1,54919 1,5454 1,46687 1,54228 1,22757 1,36181 1,41133 1,56262 1,47096 1,46422 1,4643 114,597 113,519 116,865 117,555 117,81 111,826 113,034 111,009 112,918 111,794 116,304 127,3 110,877 112,884 112,782 119,917 125,512 107,339 109,614 116,591 116,809 |
1,533 1,539 1,542 1,467 1,467 1,539 1,539 1,542 1,542 1,468 1,534 1,213 1,352 1,412 1,556 1,472 1,468 1,464 113,654 113,512 116,865 116,526 116,956 111,429 111,485 111,006 112,918 111,783 116,304 127,3 110,563 112,853 112,782 119,456 125,654 107,339 110,369 115,654 116,809 |
Сравнивая полученные результаты, оба метода имеют небольшие отклонения.
Расположение молекулы немного изменяется в зависимости от применяемого метода.
Положительный знак электростатического потенциала отображается зелёным цветом. В области неподелённых пар на атомах азота, кислорода электрический потенциал отрицательный, что отображается красным цветом.
Изменение потенциальной энергии связи С4–N5.
Исследуемая связь между атомами С4–N5. Задаём начальные величины начальная длина связи 0,97; конечная длина связи 2,97; шаг 0,1. Проведя расчёты, изобразим график изменения потенциальной энергии связи С4–N5 от величины растяжения.
Изменение потенциальной энергии углового напряжения C2-C3-С4.
Исследуем угол между атомами C2-C3-С4. Задаём начальные величины начальный угол 50; конечный 140; шаг 10. Проведя расчёты, изобразим график изменения энергии углового напряжения при изменении величины угла.
3.3 Исследование биологической активности с помощью программы PASS
В работе выполнено исследование биологической активности всех молекулярных структур с помощью программы PASS согласно методике п.2.2.
Дикаин
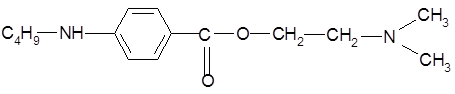
Базовые структуры лекарств, обладающие существенной новизной, целесообразно отбирать из массива доступных веществ соединения с Pa <0.7. Риск получения отрицательного результата в эксперименте тем больше, чем меньше величина Pa , однако и новизна такой структуры (при подтверждении прогноза в эксперименте) будет более высокой [12]. Pa Pi:
0.603 0.023 спазмолитик,
0.511 0.048 сосудорасширяющее средство,
0.405 0.015 антагонист кальциевых каналов,
0.350 0.107 антигипертензивный,
0.323 0.166 токсичный,
0.114 0.098 агонист β – адренорецепторов,
0.219 0.214 тератоген,
0.092 0.091 антагонист β – адренорецепторов.
1. Структура 1 (карбоксиструктура).
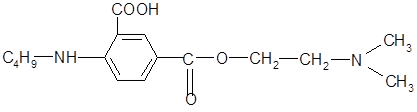 Pa Pi:
Pa Pi:
0.591 0.025 спазмолитик,
0.367 0.095 сосудорасширяющее средство,
0.264 0.051 антагонист кальциевых каналов,
0.331 0.160 токсичный,
0.301 0.142 антигипертензивный,
0.211 0.144 диуретик,
0.233 0.195 тератоген,
0.113 0.101 агонист β – адренорецепторов,
0.092 0.090 антагонист β – адренорецепторов.
2. Структура 2 (адреноструктура).
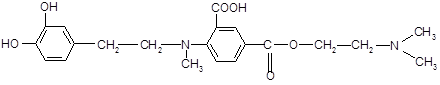 Pa Pi:
Pa Pi:
0.620 0.021 спазмолитик,
0.472 0.059 сосудорасширяющее средство,
0.362 0.020 антагонист кальциевых каналов,
0.218 0.041 агонист дофамина,
0.128 0.020 агонист Д2 дофамина,
0.291 0.188 токсичный,
0.144 0.041 агонист β1 – адренорецепторов,
0.139 0.043 агонист β – адренорецепторов,
0.243 0.182 тератоген,
0.237 0.211 антигипертензивный,
0.133 0.119 агонист α – адренорецепторов.
3. Структура 3 (никотиноструктура).

Pa Pi:
0.683 0.017 сосудорасширяющее средство,
0.548 0.031 спазмолитик,
0.326 0.026 антагонист кальциевых каналов,
0.364 0.098 антигипертензивный,
0.171 0.066 агонист дофамина.
4. Структура 4 (пиперидиноструктура).

Pa Pi:
0.680 0.015 спазмолитик,
0.537 0.042 сосудорасширяющее средство,
0.411 0.014 антагонист кальциевых каналов,
0.402 0.078 антигипертензивный,
0.233 0.051 антагонист α1 – адренорецепторов,
0.253 0.075 агонист α – адренорецепторов
0.216 0.081 антагонист адреналина.
После исследования биологической активности ряда веществ можно сделать вывод, что все структуры обладают новизной. Есть большая вероятность, что они будут обладать спазмолитической, сосудорасширяющей активностью и являются антагонистами кальциевых каналов.
Суммарно характеристики биологической активности всех молекулярных структур предложены в таблице 6.
Таблица 6
| Характерис-тика фармако-логической активности |
Основ-ная струк-тура |
Модифицированные структуры |
|||
| Дикаин |
Структура 1 Карбокси-структура |
Структура 2 Адрено-структура |
Структура 3 Никотино-структура |
Структура 4 Пиперидиноструктура |
|
| 1. Спазмолитик |
0,603 0,023 |
0,591 0,025 |
0,620 0,021 |
0,683 0,017 |
0,680 0,015 |
| 2. Сосудорасши-ряющее средство |
0,511 0,048 |
0,367 0,095 |
0,472 0,059 |
0,548 0,031 |
0,537 0,042 |
| 3. Антагонист кальциевых каналов |
0,405 0,015 |
0,264 0,051 |
0,362 0,020 |
0,326 0,026 |
0,411 0,014 |
| 4. Антигипер-тензивный |
0,350 0,107 |
0,301 0,142 |
0,237 0,211 |
0,364 0,098 |
0,402 0,078 |
| 5. Агонист β-адренорецепторов |
0,114 0,098 |
0,113 0,101 |
0,139 0,043 |
||
| 6. Токсичный |
0,323 0,166 |
0,331 0,160 |
0,291 0,188 |
||
| 7. Тератоген |
0,219 0,214 |
0,233 0,195 |
0,243 0,182 |
||
| 8. Антагонист β-адрено-рецепторов |
0,092 0,091 |
0,092 0,09 |
|||
| 9. Диуретик |
0,211 0,144 |
||||
| 10. агонист |
0,144 0,041 |
||||
| 11. Агонист α-адрено-рецепторов |
0,133 0,119 |
0,253 0,075 |
|||
| 12. Антагонист |
0,233 0,051 |
||||
Краткое описание позиций проявленной фармакологической активности.
1. Спазмолитик.
Лекарственное средство, понижающее тонус и двигательную активность гладких мышц; применяют для предупреждения или устранения спазмов гладкомышечных органов.
По механизму действия спазмолитические средства делят на миотропные и нейротропные. Миотропные спазмолитические средства снижают тонус гладкомышечных органов путем прямого влияния на биохимические процессы в гладкомышечных клетках. Нейротропные спазмолитические средства оказывают спазмолитический эффект путем нарушения передачи нервных импульсов в вегетативных ганглиях или в области окончаний вегетативных нервов, стимулирующих гладкие мышцы [19].
2. Сосудорасширяющее средство (α- и β-адреноблокаторы).
Лекарственное средство, вызывающее расширение кровеносных сосудов.
По принципу действия различают нейротропные, миотропные сосудорасширяющие средства, антагонисты кальция и сосудорасширяющие средства, влияющие на гуморальную регуляцию сосудистого тонуса.
К нейротропным сосудорасширяющим средствам относят препараты, влияющие на эффективную иннервацию сосудов [18].
3. Антагонист кальциевых каналов.
Механизм сосудорасширяющего действия препаратов группы антагонистов кальция связывают с блокадой кальциевых каналов, что приводит к затруднению проникновения ионов кальция внутрь клетки и расслаблению гладкой мускулатуры. Из числа антагонистов кальция в медицинской практике широко используется верапамил и нифедипин, которые применяют в основном как антиангинальные средства [18, 20].
4. Антигипертензивный.
Антигипертензивный – свойство вещества, препятствующего повышению гидростатического давления в полости организма, полых органах и сосудах.
Антигипертензивные вещества препятствуют развитию гипертензивного синдрома – симптомокомплекса, обусловленного стабильным или прогрессирующим поведением внутричерепного давления [8].
5. Токсичный.
Токсичность – свойство вещества синтетического и природного происхождения при поступлении в организм в количестве, превышающем меру их фармакологической активности, что выражается в возникновении токсических эффектов разной направленности, интенсивности и продолжительности вплоть до развития отравления [20].
6. Агонист β-адренорецепторов.
Агонист β-адренорецепторов – лекарственное вещество, которое прикрепляясь к β-адренорецептору, индуцирует эффективное конформационное изменение [3].
7. Тератоген.
Тератоген – фактор, вызывающий развитие врожденных пороков [5].
8. Антагонист β–адренорецепторов.
Антагонист β-адренорецепторов – лекарственное вещество, которое прикрепляется к β-адренорецептору, не индуцирует эффективного конфигурационного изменения.
β-адренолитики блокируют β-адренорецепторы, осуществляющие симпатическую иннервацию сердца (возбуждение) и торможение гладких мышц бронхов, желудка, некоторых сосудов, ресничной мышцы, поперечнополосатых мышц, а также регуляцию гликогенолиза и липолиза [7].
9. Диуретик.
Диуретики (мочегонные средства) – лекарственные средства, увеличивающие выделение почками ионов натрия и воды и вызывающие в связи с этим уменьшение содержания жидкости в тканях и серозных полостях организма.
Основным и практически важным эффектом мочегонного средства является увеличение выделения ионов натрия.
Одновременно с выделением натрия мочегонные средства способствуют выделению других ионов [19].
10. Агонист ![]() -адренорецепторов.
-адренорецепторов.
Агонист ![]() -адренорецепторов – вещество, которое посредством прикрепления к рецептору индуцирует эффективное конформационное изменение.
-адренорецепторов – вещество, которое посредством прикрепления к рецептору индуцирует эффективное конформационное изменение.
![]() -адренорецепторы опосредуют влияние катехоламинов на сердце, гладкие мышцы желудочно-кишечного тракта и, возможно, липолитический эффект КА [7].
-адренорецепторы опосредуют влияние катехоламинов на сердце, гладкие мышцы желудочно-кишечного тракта и, возможно, липолитический эффект КА [7].
11. Агонист α-адренорецепторов.
Агонист α-адренорецепторов – вещество, которое посредством прикрепления к α-адренорецептору индуцируют эффективное конформационное изменение.
α-адренорецепторы осуществляют возбуждение гладких мышц сосудов, гладких образований кожи, слизистых оболочек, органов брюшной полости, селезенки, сфинктеров желудочно-кишечного тракта и мочевого пузыря, мышцы, расширяющей зрачок и др. Сильное α-адренолитическое действие оказывают производные β-галоидоалкиламина, которые вызывают необратимую блокаду адренореактивных систем [7].
12. Агонист α-адренорецепторов.
Агонист α-адренорецепторов – вещество, которое посредством прикрепления к α-адренорецептору индуцируют эффективное конформационное изменение.
α-адренорецепторы осуществляют возбуждение гладких мышц сосудов, гладких образований кожи, слизистых оболочек, органов брюшной полости, селезенки, сфинктеров желудочно-кишечного тракта и мочевого пузыря, мышцы, расширяющей зрачок и др. Сильное α-адренолитическое действие оказывают производные β-галоидоалкиламина, которые вызывают необратимую блокаду адренореактивных систем [7].
Из таблицы 6 видно, что порог ингибирования ![]() практически для всех видов биологической активности незначителен, поэтому в дальнейшем сравнительный анализ фармакоактивности будем проводить по порогу активности
практически для всех видов биологической активности незначителен, поэтому в дальнейшем сравнительный анализ фармакоактивности будем проводить по порогу активности ![]() . Одновременно приведем значения
. Одновременно приведем значения ![]() программы PASS в условные проценты относительно базовой структуры – дикаина, принемая его характеристики за 100 %.
программы PASS в условные проценты относительно базовой структуры – дикаина, принемая его характеристики за 100 %.
Таблица 7
| Дикаин |
Карбокси-структура |
Адрено-структура |
Никотино-структура |
Пиперидно-структура |
|
| 1. Спазмолитик |
100 (0,603) |
98,00 (0,591) |
102,82 (0,620) |
113,27 (0,693) |
112,77 (0,680) |
| 2. Сосудорасширя-ющее |
100 (0,511) |
71,82 (0,367) |
92,37 (0,472) |
107,24 (0,548) |
105,09 (0,537) |
| 3. Антагонист Ca каналов |
100 (0,405) |
65,19 (0,264) |
89,38 (0,362) |
80,49 (0,326) |
101,48 (0,411) |
| 4. Антигипертен-зивный |
100 (0,350) |
86,00 (0,301) |
67,71 (0,237) |
104,00 (0,364) |
114,8 (0,402) |
| 5. Агонист β-адренорецеп-торов |
100 (0,114) |
99,12 (0,113) |
119,30 (0,139) |
||
| 6. Токсичность |
100 (0,323) |
102,48 (0,331) |
90,09 (0,291) |
||
| 7. Тератоген |
100 (0,219) |
106,39 (0,233) |
110,96 (0,243) |
||
| 8. Антагонист β-адренорецеп-торов |
100 (0,092) |
100 (0,092) |
|||
| 9. Диуретик |
(0,211) |
||||
| 10. Агонист |
(0,144) |
||||
| 11 Агонист α-адренорецеп-торов |
(0,133) |
(0,253) |
|||
| Антагонист |
(0,233) |
Сравнивая характеристики фармакологических структур и их соотношение, можно сделать следующие выводы.
1. Чем больше показатель спазмолитических свойств, тем больше анестезирующий эффект.
2. Чем меньше показатель сосудорасширяющего свойства, тем больше анестезирующий эффект.
3. Чем больше показатель антагонист кальциевых каналов, тем больше анестезирующий эффект.
4. Чем больше антигипертензивный показатель, тем меньше токсичность.
5. Чем больше показатель сосудорасширяющего средства, тем меньше токсичность.
6. Появление диуретических свойств снижает токсичность.
7. Появление α, β-антагонистов адренорецепторов уменьшает токсичность.
В нашей работе для комплексной оценки анестезирующих и токсических свойств предлагается использовать интегральные показатели.
Расчет интегральных показателей проводили по формуле 1.
 ;
;
где ![]() -интегральный коэффициент анестезирующей активности.
-интегральный коэффициент анестезирующей активности. ![]() -порог активности каждого i – вида фармакологического действия, влияющего на анестезирующий эффект.
-порог активности каждого i – вида фармакологического действия, влияющего на анестезирующий эффект. ![]() -порог активности дикаина по соответствующему виду. n
-
число видов фармакологического действия, влияющего на анестезирующий эффект.
-порог активности дикаина по соответствующему виду. n
-
число видов фармакологического действия, влияющего на анестезирующий эффект.
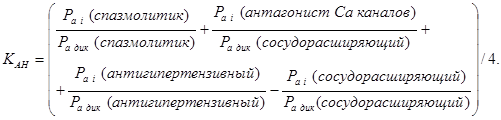
Для суммарной оценки токсических свойств предложен интегральный показатель токсичности.
 ;
;
где ![]() -интегральный коэффициент токсичности.
-интегральный коэффициент токсичности. ![]() -порог активности каждого j-вида фармакологического действия, влияющего на токсичность.
-порог активности каждого j-вида фармакологического действия, влияющего на токсичность. ![]() -порог активности дикаина по соответствующему фармакологического действия. n
-число видов фармакологического действия, влияющего на токсичность.
-порог активности дикаина по соответствующему фармакологического действия. n
-число видов фармакологического действия, влияющего на токсичность.

Таблица 8
| Интегральные коэфф-ы |
Дикаин |
Структура1 Карбокси-структура |
Структура 2 Адрено-структура |
Структура 3 Никотино-структура |
Структура 4 Пиперидино-структура |
| Коэффициент анестези-рующей активности |
0,5 |
0,443 |
0,419 |
0,543 |
0,560 |
| Коэффициент токсичности |
0,5 |
0,486 |
0,564 |
- |
- |
Из таблицы видно, что по анестезирующему эффекту исследуемые структуры можно ранжировать в следующий ряд:
Пиперидиноструктура>никотиноструктура>дикаин>
>карбоксиструктура>адреноструктура
Наглядно эффект анестезирующей активности представлен на диаграмме 1.
По токсичности исследуемые структуры располагаются в следующий ряд:
Таким образом, результаты компьютерного дизайна молекулы дикаина с целью снижения токсичности и усиления местноанестезирующего эффекта позволяют исследуемые структуры расположить в следующий ряд:
Никотиноструктура>пиперидиноструктура>адреноструктура>>дикаин>карбоксиструктура
4. Экономическая часть
4.1 Цель и база сравнения
Несмотря на достижения современной анестезии, продолжаются поиски менее опасных средств для наркоза, разработка различных вариантов многокомпонентного избирательного наркоза, позволяющего значительно снизить дозы используемых средств, уменьшить их токсичность и побочные отрицательные влияния.
В последнее время методы компьютерного моделирования все более входят в практику технологии создания новых синтетических лекарственных веществ. Полученные таким образом данные позволяют более целенаправленно проводить синтезы биоактивных молекул с заданными на молекулярном уровне параметрами, что значительно экономит время, материалы и силы при аналоговом поиске лекарственных веществ.
4.2 Проведение работы связано с определенными видами затрат
Затраты на проведение работы включают в себя:
1) Расчет заработной платы работнику, выполняющему данную работу с окладом 3500 р/мес.
2) Затраты на электроэнергию с ценой 1 кВт-1,6 р.
3) Затраты на покупку компьютера и приобретение программы HyperChem.
4.3 Заработная плата рассчитывается на 1 человека
Оклад составляет 3500 р/мес. Работа проводилась 4 месяца. Заработная плата за 4 месяца составляет 3500. 4=14000 р.
Отчисления на социальные нужды:
1) Пенсионный фонд ![]() .
.
2) Фонд социального страхования ![]() .
.
3) Фонд медицинского страхования ![]() .
.
4) Фонд страхования от несчастных случаев ![]() р.
р.
Итого: 5222 р.
Основные производственные фонды
Стоимость компьютера 20000 р.
Стоимость компьютера Hyper Chem 30000 р.
Итого: 50000 р.
Амортизация
![]()
4.4 Затраты на электроэнергию
Цена за 1 кВт – 1,6 р.
Затраты на энергоресурсы составили 0,1 кВт/ ч.
Работа на компьютере составили 528 ч.
![]()
Смета затрат
| Статьи затрат |
Стоимость, руб |
| Информационная программа HyperChem |
30000 |
| Заработная плата |
19222 |
| Амортизация |
1920 |
| Затраты на электроэнергию |
844,8 |
| ИТОГО |
51986 |
Список литературы
1. Поройков В.В. Компьютерное предсказание биологической активности веществ: пределы возможного. Химия в России, 1999, № 2, 8-12.
2. Кнунянц И. Л. Химическая энциклопедия. Издательство “Советская энциклопедия” Москва, 1988.
3. Кукес В. Г., Стародубцева А. К. Фармакология и фармакотерапия. - М.: ГЭОТАР – МЕД, 2004.
4. Беликов В. Г. Фармацевтическая химия. – М.: Высшая школа, 1985
5. Харкевич Д. А. Фармакология, четвертое издание, Москва, 1993.
6. Солдотенков А. Т., Колядина Н. М., Шендрик И. В. Основы органической химии лекарственных веществ. – М.: МИН, 2003.
7. Аляутдин Р. Н. Фармакология. – учебник для вузов, Москва, 2004.
8. Ланса Л., Лейси Ч., Голдман. М. Фармакологический справочник, Москва, 2000 г.
9. Поройков В.В., Филимонов Д.А. Компьютерный прогноз биологической активности химических соединений как основа для поиска и оптимизации базовых структур новых лекарств. В сб.: Азотистые гетероциклы и алкалоиды. Москва: Иридиум-пресс, 2001, т.1, с.123-129.
10. Poroikov V.V., Filimonov D.A., Borodina Yu.V., Lagunin A.A., Kos A. Robustness of biological activity spectra predicting by computer program PASS for non-congeneric sets of chemical compounds. J. Chem. Inform. Comput. Sci., 2000, 40 (6), 1349-1355.
11. Anzali S., Barnickel G., Cezanne B., Krug M., Filimonov D., Poroikov V. Discriminating between drugs and nondrugs by Prediction of Activity Spectra for Substances (PASS). J. Med. Chem., 2001, 4 (15), 2432-2437.
12. Лагунин А.А., Филимонов Д.А., Поройков В.В. Компьютерный поиск потенциальных антигипертензивных соединений комбинированного действия. Хим.-фарм. журн., 2001, 35 (7), 28-34.
13. Filimonov D., Poroikov V., Borodina Yu., Gloriozova T. Chemical similarity assessment through multilevel neighborhoods of atoms: definition and comparison with the other descriptors. J. Chem. Inf. Comput. Sci., 1999, 39 (4), 666-670.
14. Lagunin A., Stepanchikova A., Filimonov D., Poroikov V. PASS: prediction of activity spectra for biologically active substances. Bioinformatics, 2000, 16 (8), 747-748.
15. Poroikov V., Akimov D., Shabelnikova E., Filimonov D. Top 200 medicines: can new actions be discovered through computer-aided prediction? SAR and QSAR in Environmental Research, 2001, 12 (4), 327-344.
16. Poroikov V., Filimonov D. Computer-aided prediction of biological activity spectra. Application for finding and optimization of new leads. Rational Approaches to Drug Design, Eds. H.-D. Holtje, W.Sippl, Prous Science, Barcelona, 2001, p.403-407.
17. Кудрин А. Н. Фармакология, Москва “Медицина”, 1991.
18. Лоуренс Д. Р., Беннетт П. Н. Браун М. Дж. Фармакология. Издание второе. Москва, 2002.
19. Кудрин А. Н. Фармакология. – М.: Медицина, 2001.
20. Лоуренс Д. Р., Беннетт П. Н. Фармакология Том 1. Москва, 1993.